

Abstract
Obliterative bronchiolitis (OB) is the histopathological finding in chronic lung allograft rejection. Mounting evidence suggests that epithelial damage drives the development of airway fibrosis in OB. Tissue inhibitor of metalloproteinase (TIMP)-1 expression increases in lung allografts and is associated with the onset of allograft rejection. Furthermore, in a mouse model of OB, airway obliteration is reduced in TIMP-1-deficient mice. Matrilysin (matrix metallproteinase-7) is essential for airway epithelial repair and is required for the re-epithelialization of airway wounds by facilitating cell migration; therefore, the goal of this study was to determine whether TIMP-1 inhibits re-epithelialization through matrilysin. We found that TIMP-1 and matrilysin co-localized in the epithelium of human lungs with OB and both co-localized and co-immunoprecipitated in wounded primary airway epithelial cultures. TIMP-1-deficient cultures migrated faster, and epithelial cells spread to a greater extent compared with wild-type cultures. TIMP-1 also inhibited matrilysin-mediated cell migration and spreading in vitro. In vivo, TIMP-1 deficiency enhanced airway re-epithelialization after naphthalene injury. Furthermore, TIMP-1 and matrilysin co-localized in airway epithelial cells adjacent to the wound edge. Our data demonstrate that TIMP-1 interacts with matrix metalloproteinases and regulates matrilysin activity during airway epithelial repair. Furthermore, we speculate that TIMP-1 overexpression restricts airway re-epithelialization by inhibiting matrilysin activity, contributing to a stereotypic injury response that promotes airway fibrosis via bronchiole airway epithelial damage and obliteration.
Bronchiolitis obliterans syndrome (BOS) is the manifestation of chronic lung rejection and limits the 5-year survival after lung transplant to less than 50%.1 In comparison, transplantations of other solid organs, such as the heart, pancreas, liver, and kidneys, have 5-year survival rates exceeding 70%.2 Obliterative bronchiolitis (OB) is the histopathological equivalent of BOS and is characterized by small airway fibrosis that contributes to progressive respiratory failure and death.3 Although the pathogenesis of OB is poorly understood, evidence suggests that the primary immunological target in the lung allograft is the airway epithelium.4,5,6,7 Moreover, aberrant airway re-epithelialization is apparently sufficient for the progression of fibrosis during the allograft rejection process.8,9,10,11,12,13
The airway epithelium is an important barrier in the innate defenses of the lung.14 After lung transplantation, persistent allo-dependent (eg, acute rejection) and allo-independent (eg, infection, ischemia) pressures on the airway epithelium necessitate rapid re-epithelialization to prevent further damage that can contribute to inflammation and fibrosis.3 Disturbances in the epithelial barrier are quickly repaired through coordinated processes by which epithelial cells bordering the injury quickly spread over the denuded basement membrane.15,16,17,18 Concurrently, sheets of epithelial cells migrate over the injured area by either a sliding or leapfrog action.19,20,21 Cell proliferation is typically a later event that does not affect the initial rate of re-epithelialization in the wound.16,21
Several matrix metalloproteinases (MMPs) are selectively expressed in response to tissue injury and function in various repair processes.22 In the lungs, matrilysin (MMP-7) is induced after injury and is required for re-epithelialization of airway wounds by facilitating cell migration.23,24 Because MMPs recognize multiple substrates, their activity must be tightly regulated to ensure specificity. The tissue inhibitors of metalloproteinases (TIMP-1 through TIMP-4) are thought to function as natural MMP inhibitors. TIMPs noncovalently bind the MMP catalytic domain thereby preventing substrate proteolysis by steric hindrance. Although there is much overlap in the ability of TIMPs to inhibit MMPs in vitro, individual TIMPs appear to function in a nonredundant manner in more complex settings, such as cells or in vivo.25 In particular, TIMP-1 reduces the migratory ability of epithelial cell lines26,27 potentially by inhibiting MMP-mediated catalysis.28,29,30 However, a direct interaction between TIMP-1 and a MMP has not been demonstrated in a physiological model.
TIMP-1 expression by epithelial cells increases during epithelial regeneration in various wound-healing and disease models,31,32,33,34,35,36,37,38 including lung transplantation and during the onset of BOS.39,40,41,42 Transgenic overexpression of TIMP-1 by keratinocytes delays skin wound closure in vivo,43 suggesting that the endogenous protein functions to govern re-epithelialization. Consistent with this idea, we reported that TIMP-1 deficiency protects against chronic allograft rejection in a mouse model of OB,44 suggesting that TIMP-1 has a detrimental role in the pathogenesis of OB. However, the mechanism of how TIMP-1 functions to moderate epithelial repair has not been described. Because MMPs, specifically matrilysin, facilitate wound repair, we hypothesized that TIMP-1 restricts re-epithelialization by inhibiting the promigratory activity of this proteinase.
In the current study, we found that TIMP-1 was expressed in the airway epithelium of OB lung specimens and co-localized with matrilysin. Furthermore, we demonstrated that TIMP-1 binds matrilysin to regulate cell spreading and migration in vitro and airway re-epithelialization in vivo. Our findings suggest TIMP-1 overexpression can inhibit matrilysin to prevent re-epithelialization in lung allografts resulting in a stereotypic injury response that promotes fibroproliferation and bronchiole airway obliteration.45
Materials and Methods
Air-Liquid Interface (ALI) Cell Culture
Primary airway epithelial cells were isolated and cultured at an ALI as described.46 In brief, male wild-type (WT), matrilysin-deficient (Mat−/−)47 or TIMP-1-deficient (Timp1−/−)48 littermates all on a C57BL/6 background were euthanized, and tracheas were removed with sterile technique. Airway epithelial cells were isolated after an overnight incubation at 4°C in 1.5 mg/ml Pronase (Roche, Indianapolis, IN) and seeded in polyester transwells with 0.4-μm pore size (Corning, Acton, MA) precoated with rat tail type I collagen (BD Biosciences, Franklin Lakes, NJ). Cultures were initially grown in 5% CO2 at 37°C with medium placed in both apical and basal compartments. After reaching confluence, as determined by a transepithelial resistance greater than 1 kΩ, cultures were transitioned to an ALI with media only in the basal compartment. ALI cultures were allowed to grow and differentiate for at least 3 weeks before use.
Wound Healing Assay
After incubation in serum-free medium for 1 hour, uniform wounds were created by gently scratching a sterile P-200 pipette tip across the surface of the ALI cultures. Cultures were washed with phosphate-buffered saline (PBS) to remove cellular debris, and wounded ALI cultures were allowed to heal for 24 hours in serum-free medium. In some conditions, the medium was supplemented with 25 μmol/L of a nonspecific hydroxamate inhibitor of MMPs (MMPI) (GM6001; Millipore, Billerica, MA), 10 nmol/L mouse TIMP-1 (R&D Systems, Minneapolis, MN), or anti-murine MMP-7 blocking antibody supplied by Jason O’Neill (Amgen Inc., Seattle, WA). Phase-contrast photomicrographs of the wounds were obtained at baseline and 24 hours after injury. ALI cultures 24 hours after injury were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) in PBS, pH 7.4, overnight before changing to 70% ethanol for long-term storage.
An airway epithelial cell line (A549) was stably transfected with an autoactivating mutant of human promatrilysin (aMat8) or control vector containing either an empty plasmid or plasmid containing the nonfunctional gene, chloramphenicol acetyltransferase (A549-V1). The aMat8 cells were previously characterized to express an activated form of matrilysin.24 Confluent cells were scraped with a sterile P-200 pipette tip, washed of cellular debris, and incubated in serum-free medium alone or supplemented with 10 nmol/L human TIMP-1 or 25 μmol/L MMPI. Digital photomicrographs were obtained of the wound with a Nikon (Toyo, Japan) Diaphot phase- contrast microscope at baseline and 48 hours after injury. The wound area was quantified with the image analysis software, ImageJ (National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/). The percentage of the wound healed was calculated by the following formula:
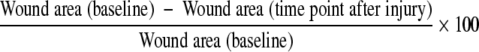 |
Cell Spreading Assay
A cell-spreading index was calculated for cells along the wound front in ALI cultures. Injured ALI cultures were hematoxylin and eosin (H&E)-stained and mounted on a glass slide. Digital photomicrographs (×100 magnification) of each wound was captured on a Nikon E600 photomicroscope with MetaMorph 4.6 software (Molecular Devices Corp., Sunnyvale, CA). The total number of nuclei entirely within a 500 μm × 500 μm square with one edge aligned to the wound front was determined with ImageJ software. The cell spreading index was calculated by the following equation:
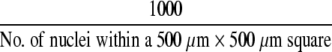 |
Cell spreading was quantified as described.49 A549-V1 or aMat8 cells were lifted from culture plates with PBS-ethylenediaminetetraacetic acid to maintain cell surface adhesive proteins. Cells were plated (2500 cells/well) into wells of a 96-well plate precoated with type IV collagen (BD Biosciences) and incubated in 100 μl of serum-free medium alone or supplemented with either 10 nmol/L human TIMP-1 or 25 μmol/L MMPI for 45 minutes at 37°C, 5% CO2, and then fixed with 4% paraformaldehyde. Phase-contrast photomicrographs were obtained from four random fields for each well. Cells were determined to display a spread morphology when they were phase dark and either surrounded by a clear ring of cytoplasm around the nucleus or elongated to greater than two times the diameter of the nucleus. Cells in all of the fields were evaluated and quantified as spread versus nonspread cells. The percentage of spread and nonspread cells were determined for each well.
Co-Immunoprecipitation and Immunoblotting
Co-immunoprecipitation (co-IP) was performed on conditioned media collected from A549-V1 cells or cell lysates of wounded ALI cultures to identify complexes of matrilysin and TIMP-1.50 After 24 hours of healing, cell lysates were collected from injured ALI cultures in ice-cold IP buffer [50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.5 mmol/L ZnCl2, 1% Brij 35 plus ethylenediaminetetraacetic acid-free protease inhibitor cocktail (Roche)]. Conditioned medium or cell lysates (200 μg total protein per condition) were precleared with 30 μl of TrueBlot anti-goat IgG IP beads (Ebioscience, San Diego, CA) for 1 hour at 4°C. IP beads were removed after centrifugation at 10,000 rpm for 5 minutes. Subsequently, either a goat anti-TIMP-1 antibody (R&D Systems) or a goat IgG control antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was added to the supernatant and incubated overnight at 4°C. Antigen-antibody complexes were precipitated with TrueBlot anti-goat IgG IP beads for 1 hour at 4°C following the manufacturer’s directions. Proteins were eluted from IP beads by heating to 95°C in reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer for 5 minutes. Samples were loaded onto a 12% Bis-Tris Gel (Invitrogen, Carlsbad, CA) and resolved by denaturing electrophoresis. Blots were incubated with a 1:1000 dilution of rabbit anti-matrilysin (Triple Point Biologics, Forest Grove, OR) in Blotto overnight at 4°C. Membranes were thoroughly washed and then incubated at room temperature for 1 hour in Blotto containing a 1:1000 dilution of horseradish peroxidase-conjugated rabbit anti-goat IgG (Ebioscience) before developing with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Naphthalene Injury Model
Male WT and Timp1−/− mice 8 to 10 weeks of age received an intraperitoneal injection of either 200 mg/kg of naphthalene (Sigma-Aldrich) or equivalent volumes of corn oil vehicle (Mazola, Cordova, TN). This dose of naphthalene has been characterized to specifically ablate Clara cells with minimal inflammation within 2 days after injection.51,52 At days 1, 2, 3, 5, and 7 after injury, mice were euthanized, and lungs were removed and instilled with 4% paraformaldehyde at an inflation pressure of 20 cm H2O. Each mouse was processed so that each paraffin-embedded tissue block contained all of the lobes of the lungs. All animals received humane care in compliance with the Principles of Laboratory Animal Care, formulated by the National Society of Medical Research, and the Guide for the Care and Use of Laboratory Animals, prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH publication no. 86-23, revised 1985). All procedures involving the mice were approved by the Fred Hutchinson Cancer Research Center Animal Studies Committee.
Immunostaining and Confocal Microscopy
Human specimens were provided by the Department of Pathology at the Fred Hutchinson Cancer Research Center. Human and mouse sections (5 μm) were cut from paraffin-embedded lungs, deparaffinized, and rehydrated. For ALI cultures, transwell membranes were cut into quarters, placed into wells of a 24-well tissue culture plate for immunostaining, and then mounted onto slides for viewing. ALI cultures and lung sections were permeabilized and washed with Tris-buffered saline, pH 7.6, plus 0.05% Tween 20 (Fisher Scientific, Pittsburgh, PA). Target Retrieval Solution (DAKO, Carpinteria, CA) was used for antigen retrieval according to the manufacturer’s instructions. Nonspecific protein binding was blocked by incubating sections with 5% goat serum in PBS (Vector Laboratories, Burlingame, CA). Sections were incubated for 1 hour at room temperature with the primary antibodies and dilutions as follows: rabbit anti-TIMP-1 (1:200, Triple Point), rabbit anti-matrilysin (1:100, Triple Point), rabbit anti-Clara cell-specific protein (CCSP) (1:1000; Upstate, Lake Placid, NY), rabbit anti-proliferating cell nuclear antigen (PCNA) (1:100; Abcam, Cambridge, MA). For negative controls, sections were processed with a rabbit control IgG antibody (Santa Cruz Biotechnology) at equivalent dilutions to the respective primary antibody.
CCSP was detected with a goat anti-rabbit IgG antibody conjugated to an Alexa 568 fluorophore (1:1000, Invitrogen). Autofluorescence was quenched with 0.3% Sudan black (Sigma-Aldrich) in 70% ethanol for 10 minutes and washed thoroughly before mounting with ProLong Gold antifade reagent (Invitrogen). All slides for CCSP immunofluorescence were processed and imaged on the same day and under identical conditions for consistency. Immunofluorescent images were captured on a Nikon E600 photomicroscope with MetaMorph 4.6 software at ×200 magnification with 1000 ms exposure time. Because naphthalene injury varies at different levels of the airways, CCSP expression was measured only in bronchioles between 150 μm and 350 μm in diameter.51,53 The fluorescent intensity for CCSP was measured on unprocessed raw data images in at least seven random airways spanning multiple lobes of the lungs in each tissue section using image analysis software (ImageJ). The fluorescent intensity was then averaged from all of the airways to produce a mean airway fluorescent intensity for each tissue specimen. Four different specimens were evaluated for each condition and time point.
When performing double immunofluorescence, sections were first labeled with matrilysin and detected with a goat anti-rabbit IgG antibody conjugated to Alexa 568 (1:1000, Invitrogen). Subsequently, sections were labeled with a TIMP-1 antibody directly conjugated with an Alexa 647 fluorophore using the Zenon antibody-labeling technology (Invitrogen). After labeling with both primary antibodies, samples were briefly fixed in 4% paraformaldehyde for 15 minutes to prevent potential dissociation and cross-contamination of secondary labeled fluorophores. Autofluorescence was quenched with 0.3% Sudan black in 70% ethanol for 10 minutes. Tissue sections were washed thoroughly before mounting with ProLong Gold antifade reagent (Invitrogen). Confocal microscopy images were obtained with the Zeiss LSM 510 Meta NLO imaging system equipped with argon, red HeNe, and green HeNe lasers mounted on a vibration-free table (Carl Zeiss, Thornwood, NY). Acquisition settings were optimized to obtain maximal signal in immunostained sections with minimal background on isotype control sections. TIFF format images for matrilysin (Alexa 568) and TIMP-1 (Alexa 647) were obtained by sequential scanning at 1024 × 1024 resolution using a × 20 objective/NA 0.75. All images were compiled with ImageJ with minor adjustments in contrast.
Statistics
Data are reported as mean ± SE. All statistical analyses were performed by the Student’s t-test and one-way analysis of variance unless otherwise noted. Differences were considered significant at the P < 0.05 level.
Results
TIMP-1 and Matrilysin Co-Localize to Epithelial Cells in OB Specimens
TIMP-1 expression is increased in the lungs after transplantation39,40,41,42 and we used immunostaining with affinity-purified antibodies to determine the cell source of its production. We identified prominent signal for TIMP-1 protein in the airway epithelium of lungs with OB (Figure 1A; and Supplemental Figure S1 at http://ajp.amjpathol.org). Matrilysin was also highly expressed by the airway epithelium in lungs from patients with OB (Figure 1B). In comparison, very low levels of TIMP-1 were detected in normal lung epithelium (data not shown) and as we reported,23 matrilysin is not detected within the intact, noninfected airway epithelium. When evaluated by confocal microscopy, signal for TIMP-1 and matrilysin proteins primarily co-localized along the obliterated airways (Figure 1C).
Figure 1.

TIMP-1 and matrilysin immunostaining in human OB tissue. A: TIMP-1 is highly expressed by the epithelium (brown, arrows) lining the obliterated airways in human OB tissue with peroxidase/DAB detection. B: Matrilysin is also expressed by the injured epithelium (brown, arrows). Serial sections showed minimal nonspecific staining with IgG control antibody or preimmune serum. C: Double immunofluorescence identifies co-localization of TIMP-1 (green, arrowheads) and matrilysin (red, arrowheads) along the damaged airways (asterisks). Original magnifications, ×200.
TIMP-1 Restrains Re-Epithelialization in Vitro via MMP Inhibition
Based on the prominence of TIMP-1 and matrilysin co-expression in the airway epithelium of OB specimens, we postulated that TIMP-1 expression in the epithelium may inhibit matrilysin-mediated airway epithelial repair. To test this hypothesis, we injured primary airway epithelial cells grown in ALI culture. These organotypic cultures differentiate into a complete mucociliary epithelium that is phenotypically similar to the airway cell composition found in vivo.46,54
Wound closure rate of Mat−/− ALI cultures was 50% of injured WT cultures (Figure 2A). Additionally, MMP inhibition with either TIMP-1 or a hydroxamate MMP inhibitor (MMPI) reduced the wound closure rate of WT cultures to that of Mat−/− ALI cultures. These data are consistent with our previous observations23,24 and confirm the necessity of matrilysin in the ALI culture model for re-epithelialization after injury. Moreover, TIMP-1 or MMPI did not further reduce the wound closure rate in Mat−/− ALI cultures (data not shown) indicating that other MMPs do not play a significant role in wound healing of injured airway epithelial cells.
Figure 2.

TIMP-1 regulates re-epithelialization in vitro. A: TIMP-1 or a nonspecific hydroxamate inhibitor of MMPs (MMPI) reduces wound closure of WT ALI cultures to the rate of injured matrilysin-deficient (Mat−/−) ALI cultures. B: Wound healing is faster in TIMP-1−/− compared to WT ALI cultures. The addition of a MMPI to TIMP-1−/− cultures abrogates this effect. C: The enhanced closure of injured TIMP-1−/− ALI cultures is reversed by the addition of inhibitory antibodies toward mouse MMP-7 (clones A6 and E12, 500 nmol/L). Control experiments were incubated with 500 nmol/L anti-streptavidin antibody (IgG-SA). The percent wound healing was calculated 24 hours after injury. Data bars represent the mean of a minimum of four experiments ± SE. *P < 0.005 by the one-way analysis of variance test. Original magnifications, ×100.
When we injured TIMP-1−/− cultures, we found that wounds closed 65% faster than WT cultures (Figure 2B). Moreover, addition of MMPI reversed the Timp1−/− phenotype by reducing the rate of wound healing to that observed in WT ALI cultures. This governing effect on wound closure appears to be specific to TIMP-1 because the rate of wound healing in Timp2−/− ALI cultures did not differ from WT cultures (data not shown). These findings indicate that TIMP-1 moderates epithelial repair through a MMP-dependent mechanism. To more specifically evaluate if the faster wound healing seen in TIMP-1−/− ALI cultures was attributable to a loss of MMP-7 inhibition, we blocked matrilysin activity in TIMP-1−/− cultures after injury using two different antibodies (clones A6 and E12) that specifically block the catalytic activity of this MMP. Inhibition of MMP-7 activity reduced wound closure rates of TIMP-1−/− cells to that of WT ALI cultures (Figure 2C).
TIMP-1 and Matrilysin Co-Localize at the Wound Front in Injured ALI Cultures
We determined the expression pattern of matrilysin and TIMP-1 in wounded ALI cultures. Consistent with our previous findings,23,24 matrilysin was prominently expressed at areas of epithelial injury (Figure 3). TIMP-1 expression was also increased at the injury front and co-localized with matrilysin in epithelial cells nearest the wound edge. Immunofluorescent staining with isotype control antibodies did not detect any nonspecific signal (data not shown). Although some groups have reported gelatinase B (MMP-9) in airway basal epithelial cells,55,56,57,58 others have not found signal for its protein or mRNA in injured or diseased airway epithelium.23,59 In our ALI culture repair model, gelatinase B expression was not detected by immunofluorescence in the wounded ALI culture (data not shown).
Figure 3.

TIMP-1 co-localizes with matrilysin at the wound front during airway epithelial repair in vitro. TIMP-1 (green) and matrilysin (red) are up-regulated in cells proximal to the wound front (arrowheads). Merged images (yellow and inset) demonstrate co-localization (arrows) of TIMP-1 and matrilysin in epithelial cells adjacent to the injury. Scale bar = 100 μm.
TIMP-1 Inhibits Matrilysin-Dependent Cell Spreading and Migration
Injured epithelium heals through a coordinated process of cell spreading and migration followed by cell proliferation.15,16,17,18 TIMP-1 does not affect the rate of epithelial cell proliferation in wounded ALI cultures as assessed by similar PCNA immunostaining in wounded WT and Timp1−/− ALI cultures (data not shown). However, close examination of the wound front in the ALI cultures revealed increased spreading by Timp1−/− cells compared to WT cells (Figure 4A). The spreading index was calculated for cells at the wound front and confirmed the observation that TIMP-1 restricts cell spreading by 50% during the re-epithelialization process. Using A549 lung epithelial cells stably transfected to overexpress active matrilysin (aMat8) or a vector control (A549-V1)24 in a cell spreading assay, we found that matrilysin facilitated cell spreading (Figure 4B). Moreover, we observed that TIMP-1 and MMPI prevented matrilysin-dependent cell spreading in aMat8 cells (Figure 4B). We also evaluated the ability of exogenous TIMP-1 to affect the migratory ability of aMat8 and A549-V1 cells in a wound healing assay (Figure 5). As reported,24 aMat8 cells migrated faster than A549-V1. Both TIMP-1 and MMPI prevented the matrilysin-mediated enhancement in cell migration. These data indicate that TIMP-1 inhibits matrilysin-mediated cell spreading and migration.
Figure 4.

Effects of TIMP-1 on cell spreading. A: Cells along the wound front are more spread (vertical bar, inset) in Timp1−/− compared to WT ALI cultures 24 hours after injury. The spreading index for each condition was calculated and confirms that Timp1−/− cells are more spread than WT cells during re-epithelialization. Bars are the mean of the spreading index in eight wounds ± SE. *P < 0.0001 by the Student’s t-test. B: Cell spreading assay using a lung epithelial cell line that overexpresses matrilysin (aMat8) compared to control (A549-V1). aMat8 cells are more spread than A549-V1 cells. TIMP-1 and a nonspecific MMP inhibitor (MMPI) abrogate the matrilysin-mediated cell spreading. Data bars represent mean of four experiments each performed in quadruplicate ± SE. *P < 0.001 by the one-way analysis of variance test. Original magnifications: ×100 (A); ×200 (B).
Figure 5.

TIMP-1 inhibits matrilysin-mediated cell migration. A lung epithelial cell line that overexpresses an autoactivating matrilysin (aMat8) or a vector control line (A549-V1) were wounded and allowed to heal. Some conditions included the addition of TIMP-1 or a nonspecific MMP inhibitor (MMPI). aMat8 cells migrate faster than A549-V1 controls in a wound healing assay. The matrilysin-mediated increase in cell migration was inhibited by both TIMP-1 and MMPI. The percent wound healing was calculated 48 hours after injury. Data bars represent the mean of at least six wounds ± SE. *P < 0.0001 by the one-way analysis of variance test. Original magnifications, ×100.
TIMP-1 Co-Immunoprecipitates with Matrilysin in Lung Wound Healing Models
The co-localization of TIMP-1 and matrilysin (Figure 3) imply that these two proteins interact during re-epithelialization. When we immunoprecipitated TIMP-1 from either conditioned medium of A549-V1 cells (Figure 6A) or from cell lysates of wounded ALI cultures (Figure 6B), matrilysin co-immunoprecipitated. Enrichment of only the active matrilysin species is consistent with the in vitro observations that TIMP-1 only binds the 19-kDa active matrilysin.60 These data support the idea that TIMP-1 binds and inhibits matrilysin at the wound front during airway re-epithelialization.
Figure 6.

Co-immunoprecipitation of matrilysin and TIMP-1 from A549-V1-conditioned medium (A) or wounded ALI culture cell lysates (B). The immunoprecipitation antibody is indicated at the top of each lane. Immunoblotting for matrilysin detects a specific band for the 19-kDa active matrilysin in the TIMP-1 IP lane. The heavy and light chains of the immunoprecipitation antibodies are detected in both lanes. The migration of molecular mass standards is shown on the right (in kDa).
TIMP-1 Deficiency Enhances Airway Re-Epithelialization in Vivo
To confirm our results with the ALI cultures, we used naphthalene injury as an in vivo model of airway epithelial repair. A single intraperitoneal injection of naphthalene selectively kills Clara cells in a dose-dependent manner within 1 day.51 We found similar degrees of airway epithelial injury in Timp1−/− and WT mice on days 1 and 2 after naphthalene injection (Figure 7A, 2, 3, 8, and 9), indicating that TIMP-1 does not influence the cytotoxicity of this compound. Corn oil vehicle produced no appreciable damage (Figure 7A, 1 and 7). At day 3 after naphthalene injury, we saw a predominance of cuboidal epithelial cells in Timp1−/− airways (Figure 7A10), whereas WT airways consisted primarily of squamous cells with visible areas of denuded basement membrane (Figure 7A4). At day 5 after injury, we observed that Timp1−/− airway epithelium contained cells morphologically resembling Clara and ciliated cells (Figure 7A11). In contrast, WT epithelium had a mixture of squamous and cuboidal cells with persistent areas of denuded basement membrane (Figure 7A5). These findings indicated that TIMP-1 restrains early stages of airway regeneration. By day 7, both WT and Timp1−/− airway epithelia had equal levels of recovery from the initial naphthalene injury (Figure 7A, 6 and 12).
Figure 7.

TIMP-1 regulates airway re-epithelialization in vivo. A: H&E-stained lung sections after naphthalene and vehicle injection. Airways are uninjured after vehicle injection (A1 and A7) with identifiable Clara (arrow) and ciliated (arrowhead) cells. One day after naphthalene injury (A2 and A8), Clara cells are vacuolated and sloughing (arrows) in both WT and Timp1−/− mice. Ciliated cells are uninjured (arrowhead). By day 2 (A3 and A9), dead cells have been cleared, and areas of denuded basement membrane are present in both WT and Timp1−/− airways (asterisks). Timp1−/− airways on day 3 after injury (A10) contain epithelial cells predominately cuboidal in appearance (arrows) whereas WT airways (A4) are primarily comprised of squamous cells (arrowheads) with identifiable areas of denuded basement membrane (asterisk). By 5 days after naphthalene injection, Clara (arrow) and ciliated (arrowhead) cells reappear in Timp1−/− airways (A11). In contrast, cuboidal (arrow) and squamous (arrowhead) cells are identified in WT airways along with persistent areas of denuded basement membrane (asterisk) (A5). Seven days after injury (A6 and A12), Clara and ciliated cells are present in both WT and Timp1−/− airways. B: Lung sections were processed and immunostained for CCSP as a marker of epithelial repair. CCSP immunofluorescence decreases equally in both genotypes around the airway lumens (asterisks) after naphthalene injury (B2, B3, B8, and B9). Airway epithelial cells in Timp1−/− mice (B10 and B11) recover CCSP staining quicker than in WT mice (B4 and B5). Naphthalene-treated airway epithefial cells at day 7 are shown in B6 and B12. Vehicle-treated controls are shown in B1 and B7. C: Quantification of mean CCSP immunofluorescence in airways of naphthalene and vehicle injected mice. Data points represent the mean CCSP immunofluorescence ± SE; arbitrary units (A.U.). n = 4 mice for all conditions. *P < 0.05 by the two-way analysis of variance test. D: Airway epithelial cell proliferation in WT and TIMP-1−/− mice after naphthalene injury. Proliferating cells were stained in lung sections with a rat polyclonal antibody against PCNA. Nuclei were identified with DAPI staining. Proliferating airway epithelial cells were counted and divided by all cells lining the airways. There was no difference in airway epithelial cell proliferation between WT and TIMP-1−/− mice after naphthalene injury. Data points represent mean percentage of PCNA-positive cells ± SE; n = 4 mice for each condition. Scale bars: 25 μm (A); 100 μm (B).
We determined the number of airway epithelial cells that stained for CCSP as a marker of airway epithelial regeneration.51 As expected for uninjured airway epithelium, we found strong expression of CCSP in the airway epithelium of WT and Timp1−/− mice injected with vehicle (Figure 7B, 1 and 7). CCSP signal was decreased 1 day after injury (Figure 7B, 2 and 8) and reached an equivalent nadir in both WT and Timp1−/− airways at day 2 after naphthalene administration (Figure 7B, 3 and 9), again indicating that TIMP-1 does not influence the extent of the initial injury. At days 3 and 5 after naphthalene injury, signal for CCSP increased in Timp1−/− airways (Figure 7B, 10 and 11) but remained at a low level in WT airways (Figure 7B, 4 and 5). By day 7 after injury, signal for CCSP increased in WT airway epithelium to levels similar to that found in Timp1−/− airways (Figure 7B, 6 and 12). Immunostaining with an isotype control antibody did not detect any signal above background (data not shown).
The intensity of CCSP immunoreactivity was measured and was significantly increased in Timp1−/− airway epithelium compared to that of the WT at days 3 and 5 after injury (Figure 7C). Alternative methods of evaluating CCSP recovering in the airways also confirmed this significant increase in CCSP positivity in TIMP-1−/− compared to WT airway epithelium at days 3 and 5 after injury (Supplemental Figure S2 at http://ajp.amjpathol.org). These findings are consistent with the morphological changes observed on H&E-stained lung sections where the repair of Timp1−/− airway epithelium was qualitatively more complete than WT epithelium at days 3 and 5 after naphthalene-induced injury (Figure 7A). These data support the concept that TIMP-1 moderates airway re-epithelialization in vivo and corroborates our cell-based data. We found no difference in the number of PCNA-positive cells between Timp1−/− and WT airway epithelia at any time suggesting that cell proliferation was not affected by TIMP-1 (Figure 7D).
TIMP-1 and Matrilysin Co-Localize in Airways after Naphthalene Injury
To localize the cell source of TIMP-1 and matrilysin expression in vivo, we examined WT tissue sections by immunofluorescence in naphthalene-injured lungs (Figure 8). TIMP-1 and matrilysin expression increased in the airway epithelium on day 3 after naphthalene injury. Moreover, TIMP-1 and matrilysin co-localized predominantly within cells adjacent to areas of denuded basement membrane. A similar pattern of TIMP-1 and matrilysin co-localization was found at days 2 and 5 with resolution to baseline expression patterns by day 7 after injury (data not shown). Immunostaining with isotype control antibodies did not detect any signal above background (data not shown). We also did not detect gelatinase B expression in the airway epithelium of naphthalene- injured lungs (data not shown).
Figure 8.

Co-localization of TIMP-1 and matrilysin in vivo during airway re-epithelialization. TIMP-1 and matrilysin are expressed minimally above background in the airway epithelium in the vehicle control section and 1 day after naphthalene injection. On day 3 after naphthalene injury, TIMP-1 and matrilysin expression increases and co-localizes (yellow) within epithelial cells adjacent to areas of denuded basement membrane (arrowheads). Scale bar = 50 μm.
Discussion
Studies have identified airway epithelial destruction as a critical event in the pathogenesis of OB.8,9,10,11,12,13 The airways of the lung allograft are constantly subjected to allo-dependent and allo-independent insults and effective repair must occur to prevent further damage. The levels of CCSP, which is expressed constitutively by Clara cells of the airway epithelium, are decreased in the bronchoalveolar lavage fluid from patients with BOS compared to normal patients, suggesting a loss of epithelial cells.61 Interestingly, CCSP can also suppress fibroblast migration.62 Additionally, stressed airway epithelial cells release profibrotic factors that can induce a remodeling response by fibroblasts,63 and delayed lung epithelial repair promotes fibroblast proliferation.64,65 Thus, evidence is mounting that epithelial damage promotes fibroproliferation. Indeed, the concept that epithelial damage promotes remodeling and fibrosis is not limited to OB and has been linked to other pulmonary diseases such as idiopathic pulmonary fibrosis and asthma66,67,68,69 as well as fibrotic diseases of the liver and kidneys.70,71,72
TIMP-1 expression increases in the lung allograft of patients with BOS/OB.39,40,41,42 Increased lung expression of TIMP-1 is also associated with the onset of OB42 and contributes to the pathogenesis of airway fibrosis in a mouse model of OB.44 Here, we find that TIMP-1 is primarily expressed by the airway epithelium in OB tissue. In addition, TIMP-1 co-localizes with matrilysin, an essential MMP for airway epithelial repair.23,24 We propose that airway epithelial overexpression of TIMP-1 hampers effective repair and could be a mechanism by which TIMP-1 contributes to the development of airway fibrosis in lung allografts. We provide direct evidence that TIMP-1 inhibits matrilysin-mediated airway re-epithelialization. We show that TIMP-1 co-localizes and co-immunoprecipitates with matrilysin from injured airway epithelial cultures. Moreover, TIMP-1 inhibits matrilysin-mediated cell migration and spreading. These cell-based results were consistent with the findings in vivo using the naphthalene injury model. Together, our data suggest that TIMP-1 coordinates matrilysin activity during re-epithelialization possibly to limit collateral damage to normal tissue. Moreover, TIMP-1 is constitutively expressed at low levels in the airway epithelium at baseline, and we speculate that TIMP-1 acts as a scavenger for MMPs in the lung airways to prevent excessive proteolysis of authentic substrates and unwarranted catalysis of potential off-target substrates.
Although TIMPs have some overlapping ability to inhibit MMPs in vitro, individual TIMPs modulate distinct biological processes in vivo as suggested by their selective expression and compartmentalization25 and by distinct phenotypes seen in null mice.44,73,74,75,76 Accordingly, we found that TIMP-1 deficiency, but not TIMP-2 deficiency, enhanced airway re-epithelialization implying that the regulation of matrilysin-mediated cell spreading and migration is specific to TIMP-1 and is not a generalized process among different members of the TIMP family. Although TIMP-1 can modulate cell apoptosis, survival, and proliferation through MMP-independent mechanisms,25,77,78 we conclude that the moderating effect TIMP-1 confers on epithelial regeneration is a consequence of its ability to block MMP activity, specifically that of matrilysin. Indeed, the phenotype was reversed when a hydroxamate MMP inhibitor or matrilysin inhibitory antibody was added to wounded Timp1−/− ALI cultures suggesting that TIMP-1 moderates re-epithelialization through MMP inhibition, specifically matrilysin.
Our data imply that overexpression of TIMP-1 impairs re-epithelialization, but we do not know what stimulates its expression in lung allografts. TIMP-1 levels are elevated in various infections and noninfectious inflammatory lung diseases.79,80,81,82,83,84,85,86 We have previously reported that TIMP-1 attenuates lung inflammation after bleomycin injury.87 Moreover, TIMP-1 neutralization augments corneal inflammation and destruction after Psuedomonas aeruginosa infection.88 Therefore, TIMP-1 may have evolved to become an important regulator of inflammation after injury and infection, similar to the proposed common function of the MMP family. However, lung transplantation is an unnatural event that creates a chronic inflammatory state. Therefore, TIMP-1 expression in the lung allograft could be increased to dampen inflammation and as an unfortunate bystander effect, restricts airway epithelial repair.
Gelatinase B (MMP-9) expression has been reported to increase in basal cells of wounded primary human bronchial epithelial cell cultures55,56,57,58 and in distal airway cells after bleomycin injury.89 However, similar to other groups,59 we have previously reported that gelatinase B was not expressed in mouse tracheas and human airways at baseline or during injury23 and once again did not find any gelatinase B expression in these current studies either in ALI cultures or in vivo. Moreover, the addition of a hydroxamate MMP inhibitor to injured Mat−/− ALI cultures had no further reduction of wound closure suggesting matrilysin is the predominate metalloproteinase needed for airway wound healing. This discordance in gelatinase B expression likely reflects differences in airway injury models.90,91,92 Nevertheless, our results show that TIMP-1 inhibits matrilysin-mediated mechanisms of re-epithelialization in models of the small diameter airways where the basal cell population is negligible.93,94 These findings are more pertinent to OB in the respect that this disease only affects the small airways.
In summary, we have demonstrated that TIMP-1 modulates matrilysin-mediated cell spreading and migration during airway re-epithelialization. Our data support the role of TIMP-1 as a physiological regulator of normal airway repair. TIMP-1 was also highly expressed by the airway epithelium in OB and co-localized with matrilysin expression. These observations, in conjunction, support the idea that TIMP-1 overexpression participate in the development of OB by pathologically inhibiting airway repair. However, our data do not rule out the possibility that TIMP-1 participates in the fibrotic process or inflammatory response in OB independent of its role in airway re-epithelialization. With these ideas in mind, we have now focused our research to further delineate the role of TIMP-1 in the pathogenesis of OB and on mechanisms by which airway epithelial damage promotes fibrosis.
Acknowledgments
We thank Paul Soloway at Cornell University (Ithaca, NY) for the generous gift of the TIMP-1−/− and TIMP-2−/− and WT breeders.
Footnotes
Address reprint requests to Peter Chen, M.D., Center for Lung Biology, 815 Mercer St., Seattle, WA 98109. E-mail: petechen@u.washington.edu.
Supported by the National Institute of Health (F32-HL079756 and K08-HL084396 to P.C.; K08-HL068780 to J.K.M.; R01-HL077555 and P01-HL029594 to W.C.P.; R01-HL63994 to D.K.M.), the American Lung Association of Washington (senior research training fellowship to P.C.), and the American Heart Association (0675038N to P.C.; 0755645Z to D.K.M.).
W.C.P. and D.K.M. contributed equally to this study.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Trulock EP, Edwards LB, Taylor DO, Boucek MM, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-third official adult lung and heart-lung transplant report–2006. J Heart Lung Transplant. 2006;25:880–892. doi: 10.1016/j.healun.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Sayegh MH, Carpenter CB. Transplantation 50 years later—progress, challenges, and promises. N Engl J Med. 2004;351:2761–2766. doi: 10.1056/NEJMon043418. [DOI] [PubMed] [Google Scholar]
- Estenne M, Hertz M. Bronchiolitis obliterans after human lung transplantation. Am J Respir Crit Care Med. 2002;166:440–444. doi: 10.1164/rccm.200201-003pp. [DOI] [PubMed] [Google Scholar]
- Mauck KA, Hosenpud JD. The bronchial epithelium: a potential allogeneic target for chronic rejection after lung transplantation. J Heart Lung Transplant. 1996;15:709–714. [PubMed] [Google Scholar]
- Tazelaar HD, Prop J, Nieuwenhuis P, Billingham ME, Wildevuur CR. Airway pathology in the transplanted rat lung. J Heart Lung Transplant. 1988;45:864–868. doi: 10.1097/00007890-198805000-00005. [DOI] [PubMed] [Google Scholar]
- Prop J, Tazelaar HD, Billingham ME. Rejection of combined heart-lung transplants in rats. Function and pathology. Am J Pathol. 1987;127:97–105. [PMC free article] [PubMed] [Google Scholar]
- Qu N, de Haan A, Harmsen MC, Kroese F, De Leij L, Prop J. Specific immune responses against airway epithelial cells in a transgenic mouse-trachea transplantation model for obliterative airway disease. Transplantation. 2003;76:1022–1028. doi: 10.1097/01.TP.0000080607.28324.A9. [DOI] [PubMed] [Google Scholar]
- Fernandez FG, Jaramillo A, Chen C, Liu DZ, Tung T, Patterson GA, Mohanakumar T. Airway epithelium is the primary target of allograft rejection in murine obliterative airway disease. Am J Transplant. 2004;4:319–325. doi: 10.1111/j.1600-6143.2004.00333.x. [DOI] [PubMed] [Google Scholar]
- Ikonen TS, Brazelton TR, Berry GJ, Shorthouse RS, Morris RE. Epithelial re-growth is associated with inhibition of obliterative airway disease in orthotopic tracheal allografts in non-immunosuppressed rats. Transplantation. 2000;70:857–863. doi: 10.1097/00007890-200009270-00002. [DOI] [PubMed] [Google Scholar]
- King MB, Pedtke AC, Levrey-Hadden HL, Hertz MI. Obliterative airway disease progresses in heterotopic airway allografts without persistent alloimmune stimulus. Transplantation. 2002;74:557–562. doi: 10.1097/00007890-200208270-00022. [DOI] [PubMed] [Google Scholar]
- Adams B, Brazelton T, Berry G, Morris R. The role of respiratory epithelium in a rat model of obliterative airway disease. Transplantation. 2000;69:661–665. doi: 10.1097/00007890-200002270-00031. [DOI] [PubMed] [Google Scholar]
- Brazelton TR, Adams BA, Cheung AC, Morris RE. Progression of obliterative airway disease occurs despite the removal of immune reactivity by retransplantation. Transplant Proc. 1997;29:2613. doi: 10.1016/s0041-1345(97)00528-9. [DOI] [PubMed] [Google Scholar]
- Genden EM, Iskander A, Bromberg JS, Mayer L. The kinetics and pattern of tracheal allograft re-epithelialization. Am J Respir Cell Mol Biol. 2003;28:673–681. doi: 10.1165/rcmb.2002-0214OC. [DOI] [PubMed] [Google Scholar]
- Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- Zahm JM, Kaplan H, Herard AL, Doriot F, Pierrot D, Somellette P, Puchelle E. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil Cytoskeleton. 1997;37:33–43. doi: 10.1002/(SICI)1097-0169(1997)37:1<33::AID-CM4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA. Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am J Physiol. 1994;267:L728–L738. doi: 10.1152/ajplung.1994.267.6.L728. [DOI] [PubMed] [Google Scholar]
- Erjefält JS, Erjefalt I, Sundler F, Persson CG. In vivo restitution of airway epithelium. Cell Tissue Res. 1995;281:305–316. doi: 10.1007/BF00583399. [DOI] [PubMed] [Google Scholar]
- Erjefält JS, Persson CG. Airway epithelial repair: breathtakingly quick and multipotentially pathogenic. Thorax. 1997;52:1010–1012. doi: 10.1136/thx.52.11.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui R, Fenteany G. Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J Cell Sci. 2005;118:51–63. doi: 10.1242/jcs.01577. [DOI] [PubMed] [Google Scholar]
- Zhao M, Song B, Pu J, Forrester JV, McCaig DC. Direct visualization of stratified epithelium reveals that wounds heal by unified sliding of cell sheets. FASEB J. 2003;17:397–406. doi: 10.1096/fj.02-0610com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk WS. A pattern of epidermal cell migration during wound healing. J Cell Biol. 1971;49:247–263. doi: 10.1083/jcb.49.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Dunsmore SE, Saarialho-Kere UK, Roby JD, Wilson CL, Matrisian LM, Welgus HG, Parks WC. Matrilysin expression and function in airway epithelium. J Clin Invest. 1998;102:1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- Roeb E, Winograd R, Breuer B, Nguyen H, Matern S. Increased TIMP-1 activity results in increased expression of gelatinases and altered cell motility. J Cell Biochem. 1999;75:346–355. [PubMed] [Google Scholar]
- Walther SE, Denhardt DT. Directed mutagenesis reveals that two histidines in tissue inhibitor of metalloproteinase-1 are each essential for the suppression of cell migration, invasion, and tumorigenicity. Cell Growth Differ. 1996;7:1579–1588. [PubMed] [Google Scholar]
- Planus E, Galiacy S, Matthay M, Laurent V, Gavrilovic J, Murphy G, Clerici C, Isabey D, Lafuma C, D’Ortho MP. Role of collagenase in mediating in vitro alveolar epithelial wound repair. J Cell Sci. 1999;112:243–252. doi: 10.1242/jcs.112.2.243. [DOI] [PubMed] [Google Scholar]
- Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;143:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Kawano Y, Tsuiki H, Sasaki J, Nakao M, Matsumoto M, Suga M, Ando M, Nakajima M, Saya H. CD44 cleavage induced by a membrane-associated metalloprotease plays a critical role in tumor cell migration. Oncogene. 1999;18:1435–1446. doi: 10.1038/sj.onc.1202447. [DOI] [PubMed] [Google Scholar]
- Coraux C, Martinella-Catusse C, Nawrocki-Raby B, Hajj R, Burlet H, Escotte S, Laplace V, Birembaut P, Puchelle E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J Pathol. 2005;206:160–169. doi: 10.1002/path.1757. [DOI] [PubMed] [Google Scholar]
- Hajj R, Lesimple P, Nowrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J Pathol. 2007;211:340–350. doi: 10.1002/path.2118. [DOI] [PubMed] [Google Scholar]
- Madlener M, Parks WC, Werner S. Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially expressed during excisional skin wound repair. Exp Cell Res. 1998;242:201–210. doi: 10.1006/excr.1998.4049. [DOI] [PubMed] [Google Scholar]
- Vaalamo M, Weckroth M, Puolakkainen P, Kere J, Saarinen P, Lauharanta J, Saarialho-Kere UK. Patterns of matrix metalloproteinase and TIMP-1 expression in chronic and normally healing human cutaneous wounds. Br J Dermatol. 1996;135:52–59. [PubMed] [Google Scholar]
- Vaalamo M, Leivo T, Saarialho-Kere U. Differential expression of tissue inhibitors of metalloproteinases (TIMP-1, -2, -3, and -4) in normal and aberrant wound healing. Hum Pathol. 1999;30:795–802. doi: 10.1016/s0046-8177(99)90140-5. [DOI] [PubMed] [Google Scholar]
- Baker EA, Leaper DJ. Proteinases, their inhibitors, and cytokine profiles in acute wound fluid. Wound Repair Regeneration. 2000;8:392–398. doi: 10.1111/j.1524-475x.2000.00392.x. [DOI] [PubMed] [Google Scholar]
- Baker EA, Leaper DJ. Profiles of matrix metalloproteinases and their tissue inhibitors in intraperitoneal drainage fluid: relationship to wound healing. Wound Repair Regeneration. 2003;11:268–274. doi: 10.1046/j.1524-475x.2003.11406.x. [DOI] [PubMed] [Google Scholar]
- Soo C, Shaw WW, Zhang X, Longaker MT, Howard EW, Ting K. Differential expression of matrix metalloproteinases and their tissue-derived inhibitors in cutaneous wound repair. Plast Reconstr Surg. 2000;105:638–647. doi: 10.1097/00006534-200002000-00024. [DOI] [PubMed] [Google Scholar]
- Xu X, Golden JA, Dolganov G, Jones KD, Donnelly S, Weaver T, Caughey GH. Transcript signatures of lymphocytic bronchitis in lung allograft biopsy specimens. J Heart Lung Transplant. 2005;24:1055–1066. doi: 10.1016/j.healun.2004.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeh KM, Beier J, Kornmann O, Micke P, Buhl R. Sputum levels of metalloproteinase-9 and tissue inhibitor of metalloproteinase-1, and their ratio correlate with airway obstruction in lung transplant recipients: relation to tumor necrosis factor-a and interleukin-10. J Heart Lung Transplant. 2001;20:1144–1151. doi: 10.1016/s1053-2498(01)00325-4. [DOI] [PubMed] [Google Scholar]
- Hübner RH, Meffert S, Mundt U, Bottcher H, Freitag S, El Mokhtari NE, Pufe T, Hirt S, Folsch UR, Bewig B. Matrix metalloproteinase-9 in bronchiolitis obliterans syndrome after lung transplantation. Eur Respir J. 2005;25:494–501. doi: 10.1183/09031936.05.00091804. [DOI] [PubMed] [Google Scholar]
- Taghavi S, Krenn K, Jaksch P, Klepetko W, Aharinejad S. Broncho-alveolar lavage matrix metalloproteases as a sensitive measure of bronchiolitis obliterans. Am J Transplant. 2005;5:1548–1552. doi: 10.1111/j.1600-6143.2005.00865.x. [DOI] [PubMed] [Google Scholar]
- Salonurmi T, Parikka M, Kontusaari S, Pirila E, Munault C, Salo T, Tryggvason K. Overexpression of TIMP-1 under the MMP-9 promoter interferes with wound healing in transgenic mice. Cell Tissue Res. 2004;315:27–37. doi: 10.1007/s00441-003-0814-1. [DOI] [PubMed] [Google Scholar]
- Chen P, Farivar AS, Mulligan MS, Madtes DK. TIMP-1 deficiency abrogates obliterative airway disease after heterotopic tracheal transplantation. Am J Respir Cell Mol Biol. 2006;34:464–472. doi: 10.1165/rcmb.2005-0344OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halloran PF, Homik J, Goes N, Lui SL, Urmson J, Ramassar V, Cockfield SM. The “injury response”: a concept linking nonspecific injury, acute rejection, and long-term transplant outcomes. Transplant Proc. 1997;29:79–81. doi: 10.1016/s0041-1345(96)00015-2. [DOI] [PubMed] [Google Scholar]
- You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol. 2002;283:L1315–L1321. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- Wilson Carole L, Heppner Kathleen J, Labosky Patricia A, Hogan Brigid LM, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloway PD, Alexander CM, Werb Z, Jaenisch R. Targeted mutagenesis of TIMP-1 reveals that lung tumor invasion is influenced by TIMP-1 genotype of the tumor but not by that of the host. Oncogene. 1996;13:2307–2314. [PubMed] [Google Scholar]
- Humphries MJ. Cell adhesion assays. Methods Mol Biol. 2000;139:279–285. doi: 10.1385/1-59259-063-2:279. [DOI] [PubMed] [Google Scholar]
- Vallon R, Muller R, Moosmayer D, Gerlach E, Angel P. The catalytic domain of activated collagenase I (MMP-1) is absolutely required for interaction with its specific inhibitor, tissue inhibitor of metalloproteinases-1 (TIMP-1). Eur J Biochem. 1997;244:81–88. doi: 10.1111/j.1432-1033.1997.00081.x. [DOI] [PubMed] [Google Scholar]
- Van Winkle LS, Buckpitt AR, Nishio SJ, Isaac JM, Plopper CG. Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am J Physiol. 1995;269:L800–L818. doi: 10.1152/ajplung.1995.269.6.L800. [DOI] [PubMed] [Google Scholar]
- Lawson GW, Van Winkle LS, Toskala E, Senior RM, Parks WC, Plopper CG. Mouse strain modulates the role of the ciliated cell in acute tracheobronchial airway injury-distal airways. Am J Pathol. 2002;160:315–327. doi: 10.1016/S0002-9440(10)64375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MJ, Phalen RF. Dosimetry implications of upper tracheobronchial airway anatomy in two mouse varieties. Anat Rec. 2002;268:59–65. doi: 10.1002/ar.10134. [DOI] [PubMed] [Google Scholar]
- Davidson DJ, Kilanowski FM, Randell SH, Sheppard DN, Dorin JR. A primary culture model of differentiated murine tracheal epithelium. Am J Physiol. 2000;279:L766–L778. doi: 10.1152/ajplung.2000.279.4.L766. [DOI] [PubMed] [Google Scholar]
- Buisson AC, Zahm JM, Polette M, Pierrot D, Bellon G, Puchelle E, Birembaut P, Tournier JM. Gelatinase B is involved in the in vitro wound repair of human respiratory epithelium. J Cell Physiol. 1996;166:413–426. doi: 10.1002/(SICI)1097-4652(199602)166:2<413::AID-JCP20>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Legrand C, Gilles C, Zahm JM, Polette M, Buisson AC, Kaplan H, Birembaut P, Tournier JM. Airway epithelial cell migration dynamics: MMP-9 role in cell-extracellular matrix remodeling. J Cell Biol. 1999;2:517–529. doi: 10.1083/jcb.146.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PM, Buhler JM, D’Ortho MP, Lebargy F, Delclaux C, Harf A, Lafuma C. Expression of matrix metalloproteinase gelatinases A and B by cultured epithelial cells from human bronchial explants. J Biol Chem. 1996;271:15580–15589. doi: 10.1074/jbc.271.26.15580. [DOI] [PubMed] [Google Scholar]
- Yao PM, Delclaux C, D’Ortho MP, Maitre B, Harf A, Lafuma C. Cell-matrix interactions modulate 92-kD gelatinase expression by human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1998;18:813–822. doi: 10.1165/ajrcmb.18.6.2984. [DOI] [PubMed] [Google Scholar]
- Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baragi VM, Fliszar CJ, Conroy MC, Ye QZ, Shipley JM, Welgus HG. Contribution of the C-terminal domain of metalloproteinases to binding by tissue inhibitor of metalloproteinases. C-terminal truncated stromelysin and matrilysin exhibit equally compromised binding affinities as compared to full-length stromelysin. J Biol Chem. 1994;269:12692–12697. [PubMed] [Google Scholar]
- Nord M, Schubert K, Cassel TN, Andersson O, Riise GC. Decreased serum and bronchoalveolar lavage levels of Clara cell secretory protein (CC16) is associated with bronchiolitis obliterans syndrome and airway neutrophilia in lung transplant recipients. Transplantation. 2002;73:1264–1269. doi: 10.1097/00007890-200204270-00013. [DOI] [PubMed] [Google Scholar]
- Lesur O, Bernard A, Arsalane K, Lauwerys R, Begin R, Cantin A, Lane D. Clara cell protein (CC-16) induces a phospholipase A2-mediated inhibition of fibroblast migration in vitro. Am J Respir Crit Care Med. 1995;152:290–297. doi: 10.1164/ajrccm.152.1.7541278. [DOI] [PubMed] [Google Scholar]
- Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc Natl Acad Sci USA. 2001;98:6180–6185. doi: 10.1073/pnas.111133298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson IY, Hedgecock C, Bowden DH. Epithelial cell-fibroblast interactions in lung injury and repair. Am J Pathol. 1990;137:385–392. [PMC free article] [PubMed] [Google Scholar]
- Adamson IY, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol. 1988;130:377–383. [PMC free article] [PubMed] [Google Scholar]
- Chapman HA. Disorders of lung matrix remodeling. J Clin Invest. 2004;113:148–157. doi: 10.1172/JCI20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchelle E, Zahm J-M, Tournier J-M, Coraux C. Airway epithelial repair. regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:726–733. doi: 10.1513/pats.200605-126SF. [DOI] [PubMed] [Google Scholar]
- Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc. 2004;1:93–98. doi: 10.1513/pats.2306034. [DOI] [PubMed] [Google Scholar]
- Jones DEJ. Pathogenesis of primary biliary cirrhosis. Gut. 2007;56:1615–1624. doi: 10.1136/gut.2007.122150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoja C, Benigni A, Remuzzi G. Cellular responses to protein overload: key event in renal disease progression. Curr Opin Nephrol Hypertens. 2004;13:31–37. doi: 10.1097/00041552-200401000-00005. [DOI] [PubMed] [Google Scholar]
- Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Dig Liver Dis. 2004;36:231–242. doi: 10.1016/j.dld.2004.01.003. [DOI] [PubMed] [Google Scholar]
- English JL, Kassiri Z, Koskivirta I, Atkinson SJ, Di Grappa M, Soloway PD, Nagase H, Vuorio E, Murphy G, Khokha R. Individual Timp deficiencies differentially impact Pro-MMP-2 activation. J Biol Chem. 2006;281:10337–10346. doi: 10.1074/jbc.M512009200. [DOI] [PubMed] [Google Scholar]
- Leco KJ, Waterhouse P, Sanchez OH, Gowing KLM, Poole AR, Wakeham A, Mak TW, Khokha R. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J Clin Invest. 2001;108:817–829. doi: 10.1172/JCI12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Soloway PD. TIMP-1 and TIMP-2 perform different functions in vivo. Ann NY Acad Sci. 1999;878:519–521. doi: 10.1111/j.1749-6632.1999.tb07714.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275:26411–26415. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Monsoor A. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Invest. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidsen ML, Wurtz SO, Romer MU, Sorensen NM, Johansen SK, Christensen IJ, Larsen JK, Offenberg H, Brunner N, Lademann U. TIMP-1 gene deficiency increases tumour cell sensitivity to chemotherapy-induced apoptosis. Br J Cancer. 2006;95:1114–1120. doi: 10.1038/sj.bjc.6603378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fligiel SE, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, Johnson KJ, Varani J. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol. 2006;37:422–430. doi: 10.1016/j.humpath.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Lanchou J, Corbel M, Tanguy M, Germain N, Boichot E, Theret N, Clement B, Lagente V, Malledant Y. Imbalance between matrix metalloproteinases (MMP-9 and MMP-2) and tissue inhibitors of metalloproteinases (TMIP-1 and TIMP-2) in acute respiratory distress syndrome patients. Crit Care Med. 2003;31:536–542. doi: 10.1097/01.CCM.0000048626.02184.F8. [DOI] [PubMed] [Google Scholar]
- Torii K, Iida K, Miyazaki Y, Saga S, Kondoh Y, Taniguchi H, Taki F, Takagi K, Matsuyama M, Suzuki R. Higher concentrations of matrix metalloproteinases in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am J Respir Crit Care Med. 1997;155:43–46. doi: 10.1164/ajrccm.155.1.9001287. [DOI] [PubMed] [Google Scholar]
- Tang LF, Du LZ, Chen ZM, Zou CC. Levels of matrix metalloproteinase-9 and its inhibitor in bronchoalveolar lavage cells of asthmatic children. Fetal Pediatr Pathol. 2006;25:1–7. doi: 10.1080/15227950600701396. [DOI] [PubMed] [Google Scholar]
- Lee KS, Jin SM, Lee H, Lee YC. Imbalance between matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in toluene diisocyanate-induced asthma. Clin Exp Allergy. 2004;34:276–284. doi: 10.1111/j.1365-2222.2004.01849.x. [DOI] [PubMed] [Google Scholar]
- Choi KH, Lee HB, Jeong MY, Rhee YK, Chung MJ, Kwak YG, Lee YC. The role of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in cryptogenic organizing pneumonia. Chest. 2002;121:1478–1485. doi: 10.1378/chest.121.5.1478. [DOI] [PubMed] [Google Scholar]
- Mautino G, Henriquet C, Jaffuel D, Bousquet J, Capony F. Tissue inhibitor of metalloproteinase-1 levels in bronchoalveolar lavage fluid from asthmatic subjects. Am J Respir Crit Care Med. 1999;160:324–330. doi: 10.1164/ajrccm.160.1.9808087. [DOI] [PubMed] [Google Scholar]
- Lemjabbar H, Gosset P, Lamblin C, Tillie I, Hartmann D, Wallaert B, Tonnel AB, Lafuma C. Contribution of 92 kDa gelatinase/type IV collagenase in bronchial inflammation during status asthmaticus. Am J Respir Crit Care Med. 1999;159:1298–1307. doi: 10.1164/ajrccm.159.4.9708080. [DOI] [PubMed] [Google Scholar]
- Kim KH, Burkhart K, Chen P, Frevert CW, Randolph-Habecker J, Hackman RC, Soloway PD, Madtes DK. Tissue inhibitor of metalloproteinase-1 deficiency amplifies acute lung injury in bleomycin exposed mice. Am J Respir Cell Mol Biol. 2005;33:271–279. doi: 10.1165/rcmb.2005-0111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernacki KA, Barrett R, Hazlett LD. Evidence for TIMP-1 protection against P. aeruginosa-induced corneal ulceration and perforation. Invest Ophthalmol Vis Sci. 1999;40:3168–3176. [PubMed] [Google Scholar]
- Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol. 2000;157:525–535. doi: 10.1016/S0002-9440(10)64563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, Shami SG, Cabral-Anderson LJ, Dekker NP. Role of nonciliated cells in renewal of the bronchial epithelium of rats exposed to NO2. Am J Pathol. 1986;123:126–133. [PMC free article] [PubMed] [Google Scholar]
- Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol. 2004;164:577–588. doi: 10.1016/S0002-9440(10)63147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am J Physiol. 2004;286:L643–L649. doi: 10.1152/ajplung.00155.2003. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Moller PC. Biology of airway basal cells. Exp Lung Res. 1991;17:513–531. doi: 10.3109/01902149109062862. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Van Winkle LS, Fanucchi MV, Plopper CG. Cellular and molecular characteristics of basal cells in airway epithelium. Exp Lung Res. 2001;27:401–415. doi: 10.1080/019021401300317125. [DOI] [PubMed] [Google Scholar]