

Abstract
The protozoan Leishmania chagasi can cause disseminated, fatal visceral leishmaniasis (VL) or asymptomatic human infection. We hypothesized that genetic factors contribute to this variable response to infection. A family study was performed in endemic neighborhoods near Natal, northeast Brazil. Subjects were assessed for VL or asymptomatic infection, defined as a positive delayed type hypersensitivity (DTH) skin test response to Leishmania antigen without disease symptoms. A genome scan of 405 microsatellite markers in 1254 subjects was analyzed for regions of linkage. The results indicated loci of potential linkage to DTH response on chromosomes 2, 13, 15 and 19, and a novel region of potential interest for VL on chromosome 9. An understanding of the genetic factors determining whether an individual will develop symptomatic or asymptomatic infection with L. chagasi may illuminate proteins essential for immune protection against this parasitic disease; findings could reveal strategies for immunotherapy or prevention.
Keywords: Visceral leishmaniasis, Delayed Type Hypersensitivity, Linkage Analysis, genetic susceptibility
Introduction
Leishmaniasis refers to a spectrum of vector-borne diseases caused by the Leishmania species protozoa. Diseases caused by different Leishmania species vary, and although there is considerable overlap, each species most often causes a specific human disease syndrome or syndromes. Visceral leishmaniasis (VL), caused by L. donovani or L. chagasi, is the cause of most leishmania-induced deaths. However, even amongst individuals infected with the same Leishmania species, there is considerable variability in clinical outcome. Environmental factors contribute but cannot account wholly for differences in outcome of infection. These observations have led scientists to examine inherent host factors that might influence the clinical expression of infection.
The current study focuses on infection with L. chagasi, the cause of VL in a highly endemic region of northeast Brazil. Infection with this protozoan can result in asymptomatic infection, detected only by a positive skin test reaction to parasite antigen, or disseminated disease with hepatosplenomegaly, fever, cachexia, and immunocompromise (1;2). The mortality rate for symptomatic VL in most endemic regions is 5-10% even when treatment is available. Most L. chagasi infections resolve spontaneously. Indeed, only about one in six infected persons develops symptomatic disease (3-5). A consistent marker of healed infection and protection against subsequent re-infection is the delayed type hypersensitivity (DTH) skin test to leishmania antigen, also called a Montenegro test (5;6). Factors that determine whether a person will spontaneously resolve L. chagasi infection and develop a DTH response, or progress to symptomatic and potentially fatal disease, are incompletely understood.
VL was previously more common in rural areas of Brazil, but since the 1980's human migration from rural to periurban areas has resulted in outbreaks in major cities in the northeast region (7-10). The existence of these regions of high exposure has enabled studies of human genetic factors that put individuals at risk for developing particularly severe infection.
The asymptomatic outcome of L. chagasi infection is particularly important, in that these individuals develop an appropriate type 1 immune response to the parasite without disease symptoms, and they are likely immune to subsequent reinfection. The goal of the current study was to locate regions in the human genome linked to development of asymptomatic L. chagasi infection in families living in endemic regiosn in northeast Brazil. Asymptomatic infection was measured by the size of induration of the Montenegro (DTH) test, which provides a quantitative measure of a type 1 DTH immune response to the parasite. Our data indicate that distinct chromosomal regions may contain genes that control the DTH response as opposed to progressive VL. This supports the hypothesis that these phenotypes represent differentially programmed immune responses after infection with the same organism.
Materials and Methods
Study area
The study site is in the eastern region of the state of Rio Grande do Norte in the peri-metropolitan area surrounding Natal, a city of about 700,000 people. Natal and the vicinity has been an endemic focus for VL since the mid-1980's (8), accounting for 70% of all reported VL cases in the state between 1990 and 2004. VL concentrates in small geographic foci, and infection rates vary over time in each endemic neighborhood. We identified neighborhoods with ongoing transmission of L. chagasi through probands with active VL admitted to public hospitals in Natal, or from medical records maintained by the National Brazilian Foundation for Health. Individuals with VL were visited during their hospital stay. If they agreed to participate, the study team visited probands and their families in their homes. Members of neighboring households were recruited for the study if the home was close, and the family size was similar to the VL case family.
Ethical considerations
Written consent was obtained from adults and parents or guardians of minors under 18 years old, and written assent was obtained from minors age 12-17. Research protocols were approved by Institutional Review Boards of the Universidade Federal do Rio Grande do Norte (numbers 19-01, and 21-01), the Comissão Nacional de Ética em Pesquisa (CONEP numbers 4581 and 4575), the University of Iowa, Johns Hopkins University, the University of Virginia and the National Human Genome Research Institute, National Institutes of Health, USA. The Brazilian Institutional Review Board is NIH approved. Subjects with medical conditions discovered during the study were treated by the study team or referred or taken to the appropriate medical resource in Natal.
Physical examination and laboratory studies
After obtaining informed consent, subjects with VL and their family members were interviewed for medical history, family relationships, and environmental exposures. Blood samples were collected for blood cell count and differential, hematocrit, leishmania serology, and selected clinical chemistries when indicated for medical reasons. DNA was extracted from blood leukocytes as described (11). Antibodies to L. chagasi were detected using an Enzyme-linked immunosorbent assay (ELISA) with Brazilian L. chagasi strain MHOM/BR/00/1669 as source of antigen (12;13). The DTH response (Montenegro test) was assessed using 25 μg of L. chagasi antigens, kindly provided by Steve Reed, PhD (Infectious Diseases Research Institute, Seattle, WA). Forty eight to 72 hours after skin test application, the area of induration was measured. Induration of ≥5 mm diameter is considered a positive response indicating prior leishmania exposure (6).
Families and environment
Families enrolled in the study included families of VL cases or nearby households, who had residing in the endemic area for over 2 years. Thus, some subjects were infected with Leishmania as determined by a positive Montenegro test, but lacked family members with a history of VL. There have been no documented cases of Leishmania spp. other than L. chagasi acquired autocthonously in the Natal region since the resurgence of VL in the 1980s. Furthermore, according to blood bank records infection with Trypanosoma cruzi, the most likely cause of a false positive Montenegro test, is lower than ** SELMA ** in this region. As such, a positive Montenegro skin test most likely indicates L. chagasi infection.
Phenotype definitions
Visceral leishmaniasis
A diagnosis of VL was verified by clinical history, laboratory validation such as a positive bone marrow smear for the parasite or positive anti-leishmania serology, and response to therapy (7). Subjects with VL either had active VL at the time of study or a validated history of active VL. Historical VL cases were validated by review of either hospital charts or the family's copies of medical records.
DTH+ (Montenegro positive)
All subjects underwent Montenegro skin testing. Although it can take between 6 months and several years to develop a Montenegro response after successful of VL, (14), the DTH+ group included only individuals with a negative history of active VL and a positive Montenegro reaction. When repeated, Montenegro tests were done more than year apart to avoid boosting (15). The Montenegro test is considered positive when induration is ≥5 mm. Montenegro size varied from 0 to 15 mm, and for this genetic study the exact mm of induration was analyzed as a quantitative trait.
Serology positive (Ab+)
This group included 56 of the 107 individuals with positive leishmania serology (Ab+) who also had a negative Montenegro response (<5 mm). For genetic analysis Ab+ individuals were coded as an unknown phenotype, since positive serology is indicative of acute infection (13), which can progress to either the VL, DTH+ or DTH− phenotypes (5;12;16).
Negative skin test and serology (DTH−)
This category included individuals with negative Leishmania serology and a DTH response (<5 mm induration) either living in a household with a VL case, or living in a family in the endemic region with at least 40% infection rate (VL and DTH+). DTH− individuals were considered likely exposed to L. chagasi, although at the first assessment they did not present evidence of immune response to the parasite.
Subjects excluded from the genetic study were three VL individuals with underlying HIV infection and four of their relatives. After these exclusions 1254 subjects were included in the genetic analyses.
Genotyping
Genotyping was performed at the Center for Inherited Disease Research (CIDR, Johns Hopkins University, Baltimore MD) under permission from the Brazilian National Research Council (RMX 18/04). Short tandem repeat (STR) markers across the human genome (n=402), a modified version of the Marshfield Genetics version 8 screening set, were genotyped on each individual. The average spacing of markers was 9cM with no gaps greater than 20cM. The overall genotype missing rate was 5.54%.
Data Quality Control
Data were released from CIDR after running Genetic Analysis System (GAS) to identify any Mendelian inconsistencies and/or systematic lab or binning problems. Relationship errors were further identified using RELCHECK (17) which examines extended pedigrees for consistent patterns of allele sharing between classes of relatives across the genome. We also identified any residual Mendelian errors using the “error” option in MERLIN (v1.0-alpha). All errors were corrected prior to analyses. Marker allele frequencies were calculated for founders only, and all markers were in Hardy-Weinberg equilibrium (HWE).
Statistical Analyses
(i) Heritability and Quantitative Linkage Analysis
The size of skin test induration in mm was log-transformed in order to normalize the data for analysis of the quantitative DTH trait. The log-transformed DTH phenotype fit a normal distribution, assessed using a quantile-quantile normal plot. All individuals classified as VL were coded as missing for DTH since the DTH size varies depending on the length of time after disease.
Familial correlations were estimated using the FCOR program of S.A.G.E. (1987; Statistical Analysis for Genetic Epidemiology v.4.8). Heritability (h2) or the proportion of variation attributable to additive genetic factors, was estimated from the sibling correlations r using the equation h2=2r.
We performed two linkage analyses on the continuous DTH phenotype using the program MERLIN (v1.0-alpha). First, we performed non-parametric quantitative regression analysis to test for allele sharing among individuals with similar DTH responses. Here the sample mean was used to estimate the phenotypic mean, and the degree of identity-by-descent (IBD) allele sharing was estimated using multipoint methods where all markers contribute information. We also performed variance components analysis using the full families in MERLIN (18). MERLIN performs variance components linkage analysis under the assumption of no dominance, and calculates marker specific heritability for each trait (19). Age and gender were included as covariates in this variance components analysis.
(ii) Qualitative Linkage Analysis
For the dichotomous disease phenotype VL, we performed non-parametric linkage analysis in Merlin. We tested for excess IBD allele sharing among affected relatives using the S-all statistic originally described by Whittemore and Halpern (20). Non-parametric allele-sharing LOD scores were also calculated using the Kong and Cox linear model (21).
For all linkage analyses, we calculated nominal p-values across the genome. For short tandem repeat (STR) markers giving suggestive evidence of linkage (p value < 0.01 for both QTL regression and variance components linkage analyses), we calculated the chromosome specific empirical p-values using gene dropping simulations in the MERLIN program. Under this simulation approach, random genotypes are generated for each marker conditional on the estimated allele frequencies, observed patterns of missing data and specified genetic map under the null hypothesis of no linkage or association to the phenotype (22). For each chromosome with a region of interest, 10,000 replicates were simulated.
Results
Study families and subjects
The study sample included 1254 subjects in 191 families. Population characteristics were similar to that previously described, and some families were included in the prior clinical report (7). The study population included members of 130 families with at least one VL case and 61 families with no VL cases (non-VL). Among the VL subjects, 78 were symptomatic at the time of study entry. Other VL individuals had a documented history of disease. VL subjects had 42.5% of their relatives infected by Leishmania, whereas non-VL families had 37.2% infection, documented by a positive Montenegro skin test reaction (≥5 mm) or positive anti-Leishmania antibodies (Table 1). The demographics and the mean size induration of the Montenegro skin test are shown in Table 2.
Table 1.
Composition of the study population by Phenotype. Pedigrees were ascertained from a hospitalized index case with visceral leishmaniasis (VL) or the nearest neighbor family (non-VL)
| Number of Pedigrees by Disease Status |
Distribution of Phenotypes | Total Subjects n |
|||
|---|---|---|---|---|---|
| VL n (%) |
DTH + n (%) |
AB + n (%) |
DTH− n (%) |
||
| VL (n=130) | 162 (18.2%) | 312 (35%) | 67 (7.5%) | 351 (39.3%) | 892 |
| Non VL (n=61) | 0 (0%) | 95 (26.2%) | 40 (11.0) | 227 (62.7%) | 362 |
| Total (n=191) | 162 (13.4%) | 407(32.5%) | 107 (8.5%) | 578 (46.1%) | 1254 |
Numbers of individuals with each phenotype are explained in the text.
Table 2.
Description of the 1254 individuals in this study population
| Age Categories in Years | ||||||
|---|---|---|---|---|---|---|
| 0-5 | 6-10 | 11-14 | >15 | Total | ||
|
Sex N(%) |
Male | 109 (18.9%) | 82 (14.2%) | 90 (15.6%) | 295 (51.2%) | 576 (45.9%) |
| Female | 117 (17.2%) | 69 (10.2%) | 106 (15.6%) | 386 (56.9%) | 678 (54.1%) | |
|
Age at VL N(%) |
Male | 59 (57.3%) | 9 (8.7%) | 7 (6.8%) | 28 (27.2%) | 103 (63.6%) |
| Female | 49 (83%) | 6 (10.2%) | 2 (3.4%) | 2 (3.4%) | 59 (36.4%) | |
|
Age at encounter N (%) |
DTH+ | 14 (3.4%) | 33 (8.1%) | 59 (14.5%) | 301 (73.9%) | 407 (32.4%) |
| VL | 88 (54.3) | 28 (17.3%) | 15 (9.25%) | 31 (19.1%) | 162 (12.9%) | |
| AB+ | 12 (11.2%) | 13 (12.1%) | 21 (19.6%) | 61 (57%) | 107 (8.5%) | |
| DTH− | 112 (19.4%) | 77 (13.3%) | 101(17.5%) | 288 (49.8%) | 578 (46.1%) | |
|
Mean Montenegro Measurement (SE) |
DTH+ | 10.1 (6.6) | 13.1 (6.5) | 14.5 (7.5) | 13.9 (7.8) | 13.8 (7.7) |
We performed a genome wide linkage scan on autosomal chromosomes for both the VL phenotype as a qualitative trait, and the size of DTH induration as a quantitative trait. The solid line in Figure 1 depicts evidence for linkage to an unobserved gene controlling risk to symptomatic VL for all chromosomes. The set of families studied included 21 concordant affected sibling pairs with VL and 161 sibling pairs discordant for VL. In addition, we had second degree relative concordant pairs including 2 avuncular pairs, plus 5 cousin pairs. There were 58 discordant cousin pairs and 93 discordant avuncular pairs for the VL phenotype. Despite our very limited number of affected relative pairs, we identified one region of interest on chromosome 9q near marker D9S1118 (p value=0.003, LOD=1.60, empirical p-value =0.0034). Figure 1 depicts the region of interest on chromosome 9.
Figure 1.
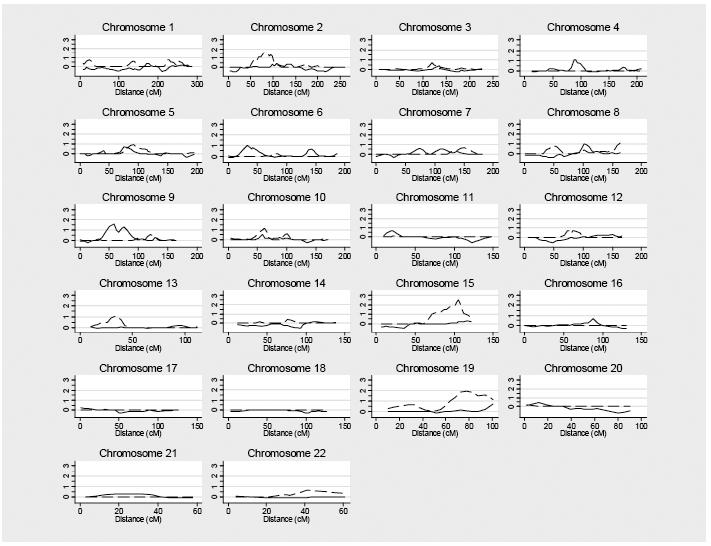
Summary of genome scan results for symptomatic visceral leishmaniasis and the size of Montenegro response. The X axis represents the distance in centiMorgans (cM) for each chromosome using the Marshfield genetic maps. Multipoint graphs of the LOD score for VL and the DTH (Montenegro) immune response. The solid line depicts the non-parametric linkage analysis of the VL phenotype. The dashed line shows results of variance components analysis of the size of Montenegro (DTH) skin test response, corrected for age and gender.
For the quantitative DTH response, there were 440 sibling pairs with a correlation of 42%, 212 grandparent-grandchild pairs with a correlation of 26.5%, and 90 cousin pairs with a correlation of 13%. This is consistent with a genetic control of DTH response, where first degree relatives have a stronger correlation than second degree relatives than third degree relatives. Estimated heritability of the DTH immune response was 84%, suggesting a substantial genetic component to variation in DTH.
There were several regions of interest for the quantitative DTH immune response trait. Dashed lines in Figure 1 depict the linkage results for the DTH immune response as measured by the (log transformed) size of the induration. We identified peak LOD scores on chromosome 15 (D15S657, VC p-value=0.0003, LOD=2.50, empirical p-value = 0.008) and chromosome 19 (D19S246, VC p-value =0.001, LOD=1.93, empirical p-value = 0.029) (Table 3). QTL LOD scores revealed a similar pattern (Table 3). This analysis also identified regions of potential interest on chromosome 2 and 13. Interestingly, when we restricted our analysis to individuals with at least one VL family member, the VC linkage peak on chromosomes 13 and 19 increased (Table 3), despite the smaller number of families. This could reflect more uniform exposure of individuals included in this latter analysis in which every family had one member with disease. Candidate genes lying within the linkage peaks are listed in Table 4.
Table 3.
Genome Wide Linkage Analysis Results of the Montenegro Response (DTH) , with corresponding analyses for Visceral Leishmaniasis in the same regions.
| Visceral Leishmaniasis |
Montenegro Response | Montenegro Response in VL families only |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Chra |
Nearest Marker |
cM |
Nonparametric Linkage Analysis 34 families |
Quantitative Regression 145 families |
Variance Components 179 families |
Quantitative Regression 98 families |
Variance Components 119 families |
|||||
| P | LOD | P | LOD | P | LOD | P | LOD | P | LOD | |||
| 2 | D2S441 | 87 | 0.5 | 0 | 0.05 | 0.56 | 0.003 | 1.60 | 0.03 | 0.73 | 0.009 | 1.21 |
| 13 | D13S894 | 33 | 0.4 | 0.02 | 0.02 | 0.99 | 0.013 | 1.09 | 0.08 | 0.44 | 0.005 | 1.43 |
| 15 | D15S657 | 105 | 0.2 | 0.18 | 0.009 | 1.22 | 0.0003 | 2.50 | 0.012 | 1.1 | 0.003 | 1.59 |
| 19 | D19S246 | 78 (73) |
0.3 | 0.07 | 0.004 | 1.54 | 0.0014 | 1.93 | 0.0012 | 2.01 | 0.0004 | 2.47 |
Chr – chromosome. cM- CentiMorgan location of the nearest marker. The number of families reflects those that were informative for linkage analysis. Both p values and LOD scores are indicated.
Table 4. VL and DTH genome scan peaks.
Shown are selected genes within the 1 LOD drop interval of VL linkage peak on chromosome 9, and the DTH linkage peaks on chromosomes 2, 13, 15 and 19. Data are based on Homo sapiens Genome Build 36.1. Genes are listed in order, with the marker position listed at the mid-position of the table.
| Chromosome 9 (VL peak D9S1118; region 21,900K-41,900K) | ||
|---|---|---|
| Symbol | Cyto | Description |
|
CDKN2A |
9p21 |
Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) |
| CDKN2B | 9p21 | Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) |
| IFNK | 9p21.2 | interferon, kappa |
| D9S1118 | ||
| ACO1 | 9p22-p13 | aconitase 1, soluble |
| TRBV20OR9-2 through TRBV29OR9-2 |
9p21 |
T cell receptor beta chain variable region genes 20-29 and A (9 genes) |
| IL11RA | 9p13 | interleukin 11 receptor, alpha |
| CCL27, CCL19, CCL21 |
9p13 | chemokine (C-C motif) ligands 27, 19 and 21 (3 genes) |
| CD72 | 9p13.3 | CD72 antigen (B cell marker) |
| Chromosome 15 (DTH peak D15S657, region 79,600,000k-telomere) | ||
| CIB1 | 15q25.3-q26 | calcium and integrin binding 1 (calmyrin) |
| MAN2A2 | 15q26.1 | Mannosidase |
| SLCO3A1 | 15q26 | solute carrier organic anion transporter family member 3A1 |
| LOC643806 | 15q26.1 | Ferritin light chain 1 homolog |
| D15S657 | ||
| IGF1R | 15q26.3 | insulin-like growth factor 1 receptor |
| Chromosome 19 (DTH peak D19S246, region 43,650K-telomere) | ||
| LGALS family | 19q13.2-q13.1 | Galectins 7, 4, 13, 14 |
| IL28B-IL28A- IL29 |
19q13.13 |
interleukins 28B, 28A, 29 |
| FCGBP | 19q13.1 | Fc γ receptor |
| TGFB1 | 19q13.1 | transforming growth factor beta 1 |
| APOE | 19q13.2 | Apolipoprotein E |
| PTGIR | 19q13.3 | Prostaglandin I2 (prostacyclin) receptor |
| CD37 | 19p13-q13.4 | CD37 |
| IRF3 | 19q13.3-q13.4 | interferon regulatory factor 3 |
| D19S246 | ||
| CD33 | 19q13.3 | CD33 (gp67) |
| CNOT3 | 19q13.4 | CCR4 subunit 3 |
| FCAR | 19q13.2-q13.4 | Fc alpha receptor |
| IL11 | 19q13.3-q13.4 | interleukin 11 |
Discussion
The outcome of Leishmania infection depends on a complex interaction between the parasite, the sand fly vector, and the mammalian host, with individual biological characteristics of each contributing to disease manifestations. This study focuses on genetic factors of the human host controlling outcome of infection with Leishmania chagasi, the cause of VL in northeast Brazil. The study population was drawn from the site of a recent resurgence of VL in periurban Natal, the largest city in the northeastern state of Rio Grande do Norte, Brazil.
The study population included 36% individuals with asymptomatic infection, 11% with symptomatic VL, and about half the subjects with no immunological evidence of L. chagasi infection. Markers of “infection” were serology, which is positive during acute infection, and the Montenegro DTH skin test, a marker of cured asymptomatic or symptomatic infection (6;13). There were DTH− subjects living in endemic households with other infected members for more than 2 years prior to entry into the study, indicating these individuals also had a high chance of exposure to the parasite. However, a small percentage of Montenegro positive individuals will revert to a negative phenotype if there is no re-exposure [(23) and unpublished data, SMBJ]. Thus, this group could include some subjects who had previously been DTH+. Due to this uncertainty, we considered only the VL phenotype and the size of DTH reaction in this linkage analysis.
This is the first genetic linkage study of asymptomatic leishmaniasis (DTH+) known to date, and our genetic linkage analysis illuminated 4 regions of interest: (chromosomes 2, 13, 15 and 19). The size of DTH reaction likely reflects the effectiveness of the type 1 immune response to L. chagasi, and DTH reaction might be a measure of development of protective immunity. These results provide intriguing information on the loci that may control the immune response to visceral leishmaniasis. The strongest evidence for linkage of the DTH response occurred on chromosomes 15 and 19, with additional smaller peaks on chromosomes 2 and 13. We hypothesize that different genes determine the propensity to develop symptomatic versus asymptomatic infection, and that multiple genes may control this immune response. The sub-analysis of VL families-only yielded higher LOD scores for two loci, suggesting unique susceptibility factors influence Montenegro responses in some families. Although none of our regions were significant using the Lander and Krugylak definition, for some loci we did have evidence of suggestive linkage ( p value = 0.00074) and these regions warrant further study (24). Highlights of good candidates genes within these linkage regions include transforming growth factor α on chromosome 2, solute anion carriers on chromosomes 2, 13, and 19, and SMAD 9 on chromosome 13. The peak marker on chromosome 19 lies close to genes encoding galectins, the Fc receptor, TGF-β1 and latent TGF-β binding protein 4, interleukin 28 subunits, and apolipoprotein E and the prostacycline receptor (Table 4). Additionally, for the subset of multiplex VL families we identified a small linkage peak for VL on chromosome 9 which lies near the IL-11 receptor α subunit, a cluster of T cell receptor beta chain variable region genes, and three chemokine receptor ligands. Further fine mapping studies are currently underway to test whether any of these candidate genes or other genes in these linkage regions control the cutaneous immune response to leishmania or the development of VL.
There are two reported genome wide linkage scans of VL to date. The first focused on residents of a Sudanese village exposed to L. donovani. This study described significant evidence for linkage of symptomatic disease to a region of chromosome 22q12 (LOD score 3.50) in a genome wide linkage scan of VL in multiplex families from a single village in the Sudan. A minor locus (22q22-q23) was also suggestive in families with negative LOD scores for the 22q12 locus (25). Curiously, other studies of individuals with VL and post-kala-azar-dermal-leishmaniasis (PKDL) in a geographically close region of the Sudan found no evidence of association with these same markers, but rather found association of VL with the IL4 and SLC11A1 genes encoding IL-4 and NRAMP1, respectively, and association of PKDL with the gene encoding the α chain of the IFN-γ receptor (IFNGR1) (26). A genome scan of VL in multiplex pedigrees from northern Brazil identified three chromosomal regions of interest (6q27, 7q11.22, and 17q11.2-q21.3), with peak LOD scores of 1.08, 1.34, and 1.14, respectively. Fine mapping in the chromosomal 17 region suggested allelic association with two chemokine genes (27). A subset of our VL families was included in that study. The current study, in which we focused on the DTH phenotype but also analyzed VL, did not replicate either the previous findings of linkage to the VL phenotype in the Brazilian or the Sudanese populations. Differences from the Sudanese study could be due to the different species of Leishmania (L. donovani in Sudan versus L. chagasi in Brazil) and genetic differences between the Sudanese and Brazilian populations. The different outcome of our study versus the other genome scan of multiplex VL families in Brazil could also reflect genetic differences between hosts, or more likely the genetic heterogeneity of the VL trait. Indeed, Brazilians are a genetically admixed population, and regional differences have already been documented between residents of different Brazilian states (28;29). It is additionally likely that the relatively small numbers of informative multiplex VL pedigrees in our study could have resulted in failing to detect all genes controlling risk to VL in endemic populations.
In summary, we report the results of our initial genome-wide scan for the DTH immune response and identify loci of interest on chromosomes 2, 13, 15 and 19. We also identified a novel region of interest for VL on chromosome 9. Further fine mapping studies in our own cohort, and replication in other populations exposed to L. chagasi, are next steps toward the goal of identifying the genes that control the host immune response to leishmaniasis. An understanding of the genetic factors determining whether an individual will develop symptomatic or asymptomatic infection by L. chagasi will help in understanding immune protection against disease caused by this parasite, and hopefully direct future efforts at immunotherapy or prevention.
Acknowledgments
Funding: The results of this article were obtained by using the program package SAGE, which is supported by the US Public Health Service Resource Grant RR03665 from the National Center for Research Resources, Bethesda, MD. This research was supported by National Institutes of Health grants AI048822 (MEW), AI45540 (MEW), NIH TMRC grant AI30639 (SMBJ, ), Merit Review and Gulf War grants from the Department of Veterans' Affairs (MEW), and in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Abbreviations
- DTH
delayed type hypersensitivity skin test
- IBD
Identical by descent
- VL
visceral leishmaniasis
Footnotes
Conflict of Interest Statement: The authors do not have a commercial or other association that might pose a conflict of interest with this work.
References
- 1.Wilson ME, Jeronimo SMB, Pearson RD. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog. 2005;38:147–60. doi: 10.1016/j.micpath.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Kaye PM. The Immunology of Visceral leishmaniasis: Current status. In: Farrell JP, editor. Leishmania. Boston: Kluwer Academic Publishers; 2002. pp. 137–50. [Google Scholar]
- 3.Badaro R, Jones TC, Lorenco R, et al. A prospective study of visceral leishmaniasis in an endemic area of Brazil. J Infect Dis. 1986;154:639–49. doi: 10.1093/infdis/154.4.639. [DOI] [PubMed] [Google Scholar]
- 4.Badaro R, Jones TC, Carvalho EM, et al. New perspectives on a subclinical form of visceral leishmaniasis. J Infect Dis. 1986;154:1003–11. doi: 10.1093/infdis/154.6.1003. [DOI] [PubMed] [Google Scholar]
- 5.Jeronimo SMB, Teixeira MJ, Sousa AdQ, Thielking P, Pearson RD, Evans TG. Natural History of Leishmania (Leishmania) chagasi infection in northeastern Brazil: long term follow-up. Clin Infect Dis. 2000;30:608–9. doi: 10.1086/313697. [DOI] [PubMed] [Google Scholar]
- 6.Reed SG, Badaro R, Masur H, et al. Selection of a skin test antigen for American visceral leishmaniasis. Am J Trop Med Hyg. 1986;35(1):79–85. doi: 10.4269/ajtmh.1986.35.79. [DOI] [PubMed] [Google Scholar]
- 7.Jeronimo SMB, Duggal P, Braz RFS, et al. An emerging peri-urban pattern of infection with Leishmania chagasi, the protozoan causing visceral leishmaniasis in northeast Brazil. Scand J Infect Dis. 2004;36(6/7):443–9. doi: 10.1080/00365540410020451. [DOI] [PubMed] [Google Scholar]
- 8.Jeronimo SMB, Oliveira RM, Mackay S, et al. An urban outbreak of visceral leishmaniasis in Natal, Brazil. Trans R Soc Trop Med Hyg. 1994;88:386–8. doi: 10.1016/0035-9203(94)90393-x. [DOI] [PubMed] [Google Scholar]
- 9.Costa CH, Pereira HF, Araujo MV. Epidemia de leishmaniose visceral no estado do Piauí, Brasil. Rev Saude Publica. 1990;24:361–72. doi: 10.1590/s0034-89101990000500003. [DOI] [PubMed] [Google Scholar]
- 10.Werneck GL, Rodrigues L, Santos MV, et al. The burden of Leishmania chagasi infection during an urban outbreak of visceral leishmaniasis in Brazil. Acta Trop. 2002;83(1):13–8. doi: 10.1016/s0001-706x(02)00058-x. [DOI] [PubMed] [Google Scholar]
- 11.Karplus TM, Jeronimo SMB, Chang H, et al. An association between the TNF locus and the clinical outcome of Leishmania chagasi infection. Infect Immun. 2002;70:6919–25. doi: 10.1128/IAI.70.12.6919-6925.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans TG, Teixeira MJ, McAuliffe IT, et al. Epidemiology of visceral leishmaniasis in northeast Brazil. J Infect Dis. 1992;166:1124–32. doi: 10.1093/infdis/166.5.1124. [DOI] [PubMed] [Google Scholar]
- 13.Braz RFS, Nascimento ET, Martins DRA, et al. The sensitivity and specificity of Leishmania chagasi recombinant K39 antigen in the diagnosis of American visceral leishmaniasis and in differentiating active from subclinical infection. Am J Trop Med Hyg. 2002;67:344–8. doi: 10.4269/ajtmh.2002.67.344. [DOI] [PubMed] [Google Scholar]
- 14.Miles SA, Conrad SM, Alves RG, Jeronimo SMB, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med. 2005;201:747–54. doi: 10.1084/jem.20041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jose FF, da Silva IM, Araujo MI, Almeida RP, Bacellar O, Carvalho EM. Evaluation of the sensitization power of Montenegro skin test. Rev Soc Braz Med Trop. 2001;34(6):537–42. doi: 10.1590/s0037-86822001000600007. [DOI] [PubMed] [Google Scholar]
- 16.Evans TG, Teixeira J, Sousa AD, Pearson RD. Short report: Extended follow-up of the natural history of persons infected with Leishmania chagasi. Am J Trop Med Hyg. 1995;53:360–1. doi: 10.4269/ajtmh.1995.53.360. [DOI] [PubMed] [Google Scholar]
- 17.Ulczak OM, Ghadirian E, Skamene E, Blackwell JM, Kongshavn PAL. Characterization of protective T cells in the acquired response to Leishmania donovani in genetically determined cure (H-2b) and noncure (H-2d) mouse strains. Infect Immun. 1989;57:2892–9. doi: 10.1128/iai.57.9.2892-2899.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin - Rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 19.Amos C. Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet. 1994;54:534–43. [PMC free article] [PubMed] [Google Scholar]
- 20.Whittemore AS, Halpern J. Probability of gene identity by descent: computation and applications. Biometrics. 2006;50(1):109–17. [PubMed] [Google Scholar]
- 21.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–88. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawcer S, Jones HB, Judge D, et al. Empirical genome wide significance levels established by whole genome simulations. Genet Epidemiol. 1997;14:223–9. doi: 10.1002/(SICI)1098-2272(1997)14:3<223::AID-GEPI1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Ali A, Ashford RW. Visceral leishmaniasis in Ethiopia. II. Annual leishmanin transformation in a population. Is positive leishmanin reaction a life-long phenomenon? Ann Trop Med Parasitol. 1993;87(2):163–7. doi: 10.1080/00034983.1993.11812750. [DOI] [PubMed] [Google Scholar]
- 24.Lander E, Krugylak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genet. 1995;11:241–7. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 25.Bucheton B, Abel L, El-Safi S, et al. A major susceptibility locus on chromosome 22q12 plays a critical role in the control of kala-azar. Am J Hum Genet. 2003;73:1052–60. doi: 10.1086/379084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohamed H, Ibrahim ME, Miller EN, et al. Genetic susceptibilty to visceral leishmaniasis in The Sudan: Linkage and association with IL4 and IFNGR1. Genes Immun. 2003;4(5):351–5. doi: 10.1038/sj.gene.6363977. [DOI] [PubMed] [Google Scholar]
- 27.Jamieson SE, Miller EN, Peacock CS, et al. Genome-wide scan for visceral leishmaniasis susceptibility genes in Brazil. Genes Immun. 2006 Nov. doi: 10.1038/sj.gene.6364357. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohlrausch FB, Callegari-Jacques SM, Tsuneto LT, et al. Geography influences microsatellite polymorphism diversity in Amerindians. Am J Phys Anthropol. 2005;126(4):463–70. doi: 10.1002/ajpa.20042. [DOI] [PubMed] [Google Scholar]
- 29.Abe-Sandes K, Silva WA, Jr., Zago MA. Heterogeneity of the Y chromosome in Afro-Brazilian populations. Hum Biol. 2004;76(1):77–86. doi: 10.1353/hub.2004.0014. [DOI] [PubMed] [Google Scholar]