

The crystal structure of MtSK in complex with ADP shows conformational changes caused by the absence of the shikimate molecule, and the effect of chloride ions on the nucleotide binding site, and the crystal structure of MtSK in absence of magnesium shows its effect on the shikimate binding site.
Keywords: shikimate
Abstract
Bacteria, fungi and plants can convert carbohydrate and phosphoenolpyruvate into chorismate, which is the precursor of various aromatic compounds. The seven enzymes of the shikimate pathway are responsible for this conversion. Shikimate kinase (SK) is the fifth enzyme in this pathway and converts shikimate to shikimate-3-phosphate. In this work, the conformational changes that occur on binding of shikimate, magnesium and chloride ions to SK from Mycobacterium tuberculosis (MtSK) are described. It was observed that both ions and shikimate influence the conformation of residues of the active site of MtSK. Magnesium influences the conformation of the shikimate hydroxyl groups and the position of the side chains of some of the residues of the active site. Chloride seems to influence the affinity of ADP and its position in the active site and the opening length of the LID domain. Shikimate binding causes a closing of the LID domain and also seems to influence the crystallographic packing of SK. The results shown here could be useful for understanding the catalytic mechanism of SK and the role of ions in the activity of this protein.
1. Introduction
Mycobacterium tuberculosis, the aetiological agent of tuberculosis (TB), infects one-third of the world’s population. It is estimated that 1.7 million deaths resulted from TB in 2004, 95% of which occurred in developing countries (World Health Organization, 2006 ▶). The emergence of TB as a public health threat, the high susceptibility of HIV/TB co-infected patients and the proliferation of multi-drug-resistant strains have created a need for newer and better drugs for the treatment of TB.
The shikimate pathway is an attractive target for the development of herbicides (Coggins, 1998 ▶) and antibiotic agents (Davies et al., 1994 ▶) because it is essential in algae, higher plants, bacteria, fungi and apicomplexan parasites but is absent from mammals (Bentley, 1990 ▶; Roberts et al., 1999 ▶). The shikimate pathway is a seven-step biosynthetic route that links the metabolism of carbohydrates to the synthesis of aromatic amino acids. The shikimate pathway leads to the biosynthesis of chorismate, which is a precursor of aromatic amino acids and many other aromatic compounds (Ratledge, 1982 ▶). Shikimate kinase (SK; EC 2.7.1.71), the fifth enzyme of this pathway, catalyzes phosphate transfer from ATP to the carbon-3-hydroxyl group of shikimate, forming shikimate 3-phosphate (S3P).
SK belongs to the nucleoside monophosphate (NMP) kinase structural family. SK is a α/β protein consisting of a central sheet of five parallel β-strands flanked by α-helices, with overall topology similar to that of adenylate kinase (Pereira et al., 2004 ▶; Krell et al., 1998 ▶, 2001 ▶). A characteristic feature of the NMP kinases is that they undergo large conformational changes during catalysis (Vonrhein et al., 1995 ▶). The NMP kinases are composed of three domains: the CORE, which contains a highly conserved phosphate-binding loop (P-loop), the LID domain, which undergoes substantial conformational changes upon substrate binding, and the NMP-binding domain, which is responsible for the recognition and binding of a specific substrate (Gu et al., 2002 ▶).
MgADP induces concerted hinged movements of the shikimate-binding (SB) and LID domains such that the two domains move towards each other and towards the active centre of the enzyme in the presence of this ligand (Gu et al., 2002 ▶).
The precise mode of binding of shikimate to MtSK and some conformational changes upon shikimate binding to MtSK have recently been proposed (Pereira et al., 2004 ▶; Dhaliwal et al., 2004 ▶). The binding of shikimate to MtSK causes a concerted conformational change of the LID and SB domains towards each other and results in an additional closure of the active site.
Chloride ions have been shown to weaken the interaction between shikimate and SK from Erwinia chrysanthemi and to strengthen the affinity of the enzyme for ADP and ATP (Cerasoli et al., 2003 ▶). Thus, a chloride ion seems to occupy a site crucial for the binding of the nucleotide substrate in the correct orientation for catalysis.
In this work, we report two crystallographic structures: the MtSK–ADP–shikimate and MtSK–MgADP complexes. The MtSK–ADP–shikimate complex has been solved at 1.93 Å resolution. The crystal structure of MtSK in complex with ADP and Mg2+ has been solved at 2.8 Å resolution. The data presented here provide an evaluation of the effects of both Mg2+ and Cl− ions and shikimate on the structure of MtSK. Thus, these results provide further insight into the roles that different ions and substrate play in the mechanism of action of MtSK.
2. Materials and methods
2.1. Crystallization
The cloning, expression and purification of MtSK have been reported elsewhere (Oliveira et al., 2001 ▶). Initially, MtSK was concentrated to 14 mg ml−1 and dialyzed against 50 mM Tris–HCl buffer pH 8.0. For the crystallization of MtSK in complex with ADP and shikimate in the absence of Mg2+ ion, variations of the crystallization condition established by Dhaliwal et al. (2004 ▶) were used. Crystals were obtained by the hanging-drop vapour-diffusion method. The well solution contained 0.1 M Tris–HCl buffer pH 8.0, 17% PEG 1500 and 0.5–0.7 M LiCl. The drops had a protein solution:well solution volume ratio of 1.6:1.0. For the crystallization of the protein in complex with ADP and Mg2+ in the absence of shikimate, a crystallization solution composed of 0.1 M Tris–HCl buffer pH 8.0, 20% PEG 3350 and 0.1 M MgCl2.6H2O was used. The concentration of the protein was 17 mg ml−1 and the drops again had a protein solution:well solution volume ratio of 1.6:1.0.
2.2. Data collection and processing
All data sets were collected at a wavelength of 1.427 Å using a synchrotron-radiation source (Station PCr, LNLS, Campinas, Brazil; Polikarpov et al., 1998 ▶) and a CCD detector (MAR CCD). Data collection was performed under cryogenic conditions at a temperature of 100 K in a cold nitrogen stream generated and maintained with an Oxford Cryosystem. Prior to flash-cooling, glycerol was added to the crystallization drop to 20%(v/v). The data sets were processed using the program MOSFLM (Leslie, 1992 ▶) and scaled with SCALA (Collaborative Computational Project, Number 4, 1994 ▶).
2.3. Structure determination
The crystal structures of both complexes were determined by standard molecular-replacement methods using the program AMoRe (Navaza, 2001 ▶). For both complexes, we used as a search model the structure of MtSK–MgADP–shikimate (PDB code 1we2; Pereira et al., 2004 ▶). Refinement of the structures was performed using REFMAC5 implemented in the CCP4 package (Murshudov et al., 1997 ▶; Collaborative Computational Project, Number 4, 1994 ▶). XtalView/Xfit (McRee, 1999 ▶) was used for visual inspection and addition of water molecules. The stereochemical correctness of the models was checked using PROCHECK (Laskowski et al., 1993 ▶). The final atomic models were superposed using LSQKAB from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). PARMODEL (Uchôa et al., 2004 ▶) was used in the final analysis of the model.
3. Results and discussion
MtSK crystallized in two different space groups depending on the complex. The crystals of the MtSK–ADP–shikimate ternary complex were trigonal, space group P3221, and diffracted to 1.93 Å resolution. The asymmetric unit contains one molecule and the final values of R and R free were 20.2 and 27.0%, respectively. In contrast, the crystals of the MtSK–ADP–Mg2+ ternary complex were orthorhombic, space group P212121, and diffracted to 2.8 Å resolution. The asymmetric unit contains four MtSK monomers that form a tetramer and the final values of R and R free were 18.3 and 28.0%, respectively. Table 1 ▶ details the data processing, refinement statistics and quality analysis of the two complexes.
Table 1. Crystallographic data, refinement statistics and analysis of the quality of MtSK structures.
Values in parentheses are for the outermost shell.
| MtSK–ADP–shikimate | MtSK–MgADP | |
|---|---|---|
| Crystallographic data | ||
| Unit-cell parameters | ||
| a (Å) | 63.3 | 60.2 |
| b (Å) | 63.3 | 62.2 |
| c (Å) | 91.6 | 170.6 |
| Space group | P3221 | P212121 |
| No. of measurements | 76792 | 97059 |
| No. of independent reflections | 16017 | 17057 |
| Completeness (%) | 96.9 (91.5) | 99.4 (96.9) |
| Rsym† (%) | 8.9 (58.8) | 12.1 (58.0) |
| Redundancy | 4.8 | 5.7 |
| Refinement statistics | ||
| Resolution range (Å) | 35.16–1.93 | 57.17–2.80 |
| Reflections used for refinement | 15130 | 15670 |
| Final R factor‡ (¨%) | 20.2 | 18.3 |
| Final Rfree§ (%) | 27.0 | 28.0 |
| Correlation coefficient (%) | 95.2 | 94.3 |
| B values (Å2) | ||
| Main chain | 31 | 33 |
| Side chain | 34 | 36 |
| ADP | 21 | 27 |
| Shikimate | 32 | — |
| Waters | 39 | 30 |
| Quality of structure | ||
| Three-dimensional profile¶ | S = 88.07, IS = 74.95, S/IS = 1.18IS | S = 343.59, IS = 294.34, S/IS = 1.17IS |
| Ramachandran plot | ||
| Favoured | 95.6 | 84.8 |
| Additionally allowed | 2.9 | 13.3 |
| Generously allowed | 0.7 | 0.8 |
| Disallowed | 0.7 | 1.1 |
R
sym = 100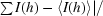
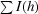 , where I (h) is the observed intensity and 〈I(h)〉 is the mean intensity of reflection h over all measurements of I(h).
, where I (h) is the observed intensity and 〈I(h)〉 is the mean intensity of reflection h over all measurements of I(h).
R factor = 100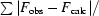
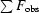 , the sums being taken over all reflections with F/σ(F) > 2σ(F).
, the sums being taken over all reflections with F/σ(F) > 2σ(F).
R free is the R factor for 10% of the data that were not included during crystallographic refinement.
The ideal score measures the compatibility of a protein model with its sequence, using a 3D profile. Each residue position in the 3D model is characterized by its environment and is represented by a row of 20 numbers in the profile. These numbers are the statistical preferences (called 3D-1D scores) of each of the 20 amino acids for this environment. Environments of residues are defined by three parameters: the area of the residue that is buried; the fraction of side chain area that is covered by polar atoms (O and N) and the local secondary structure. The 3D profile score S for the compatibility of the sequence with the model is the sum, over all residue positions, of the 3D-1D scores for the amino-acid sequence of the protein. For 3D protein models known to be correct, the 3D profile score S for the amino-acid sequence of the model is high, by contrast, the profile score S for the compatibility of a wrong 3D protein model with its sequence is often low. When this method is used to verify a structure, the raw compatibility score alone is difficult to interpret. In this case it is necessary to compare the score to those obtained using structures known to be correct, we use the Ideal Score (IS), that is calculated from the length of the protein. The IS is determined by 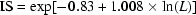 . Where L is the length of the sequence. Severely misfolded structures typically have scores less than 0.45 IS. A score near or above IS indicates a reliable structure.
. Where L is the length of the sequence. Severely misfolded structures typically have scores less than 0.45 IS. A score near or above IS indicates a reliable structure.
The structures present good geometry, although some residues of the LID domain are located in disallowed regions of the Ramachandran plot. The N-terminal methionine residue is not observed since it was removed during the MtSK expression in E. coli (Oliveira et al., 2001 ▶). The ten C-terminal residues are disordered in both structures and have not been included in the final structure.
The folding of the MtSK complexes presented here is similar to that of those reported previously (Pereira et al., 2004 ▶; Krell et al., 1998 ▶; Gu et al., 2002 ▶; Dhaliwal et al., 2004 ▶). SK is a α/β protein and consists of five central parallel β-strands flanked by α-helices. Fig. 1 ▶(a) shows a ribbon representation of the secondary-structure elements of MtSK–ADP–shikimate at 1.93 Å resolution.
Figure 1.

(a) Structure of MtSK in complex with ADP, shikimate and Cl−. (b) Tetrameric structure of MtSK in complex with ADP, Mg2+ and Cl−. The monomers A, B, C and D are represented in green, blue, pink and yellow, respectively. The dark blue and yellow spheres represent the magnesium and chloride ions, respectively. The ADP molecules are represented as sticks. (c) Representation of the hydrogen-bonding interactions that occur between the ribose hydroxyl groups of ADP molecules bound to monomers A and B. The distances are shown in Å. Figures were generated with the program MolMol (Koradi et al., 1996 ▶).
3.1. Structure of the MtSK–ADP–Mg2+ ternary complex
The asymmetric unit of the MtSK–ADP–Mg2+ ternary complex structure contains four monomers forming a homotetramer with 222 symmetry. However, each subunit presents a different conformation state. In the A and C subunits the LID domain is disordered and residues 115–123 and 114–123 were thus not included in the final model.
The structure of the MtSK tetramer is shown in Fig. 1 ▶(b). Each monomer is in contact with the other three, creating an intricate packing arrangement. The ADP molecules appear to play an important role in the stabilization of the tetramer, since the hydroxyl groups of the ribose moiety of ADP of one monomer form hydrogen bonds with those of a neighbouring monomer (Fig. 1 ▶ c).
3.2. Influence of Mg2+ on the structure of MtSK
The absence of Mg2+ ions seems to have a significant effect on the position of shikimate and some of the active-site residues, mainly Asp32 and Asp34 (these are conserved residues in all SKs and are involved in the binding of Mg2+; Fig. 2 ▶). In addition, the chloride ion and ADP molecule also undergo changes in position. Fig. 2 ▶ shows a superposition of the MtSK–MgADP–shikimate (Pereira et al., 2004 ▶) and MtSK–ADP–shikimate complexes, showing the structural changes caused by the absence of the Mg2+ ion.
Figure 2.

Superposition of the structures of the MtSK–MgADP–shikimate and MtSK–ADP–shikimate ternary complexes. The Cα trace of the MtSK–MgADP–shikimate complex is presented in green and that of the MtSK–ADP–shikimate trace is presented in blue. The C atoms of MtSK–MgADP–shikimate and MtSK–ADP–shikimate are coloured white and yellow, respectively. The Mg2+ shown in yellow and the chloride ion shown in turquoise refer to the MtSK–MgADP–shikimate structure, while the chloride ion in dark blue refers to the MtSK–ADP–shikimate structure. The figure was generated with the program MolMol (Koradi et al., 1996 ▶).
In the structure of the MtSK–ADP–shikimate ternary complex, the 3-, 4- and 5-hydroxyl groups of shikimate undergo a shift in their positions, leading to small differences in the hydrogen-bonding pattern between shikimate and MtSK. Although the Asp34 residue interacts with shikimate in both structures, a hydrogen bond between the OD2 atom of Asp34 and the O3 atom of the shikimate is not observed in the MtSK structure without Mg2+ (not shown).
The position of the chloride ion is closer to shikimate in the MtSK–ADP–shikimate ternary complex than in the MtSK–MgADP–shikimate complex (Pereira et al., 2004 ▶). In the structure of the MtSK–ADP–shikimate ternary complex, the chloride ion is 2.4 Å away from the O1 atom of the shikimate 3-hydroxyl group, while in the structure of MtSK–MgADP–shikimate the distance between these two atoms is 3.4 Å. The absence of the Mg2+ ion can also cause alterations in the side chains of the hydrophobic residues Phe49 and Phe57 located in the SB domain and of Pro118, Ala46 and Ile45 located in the LID domain (Fig. 2 ▶).
As established previously for other NMP kinases, the Mg2+ ion is expected to play an important role in the transfer of the γ-phosphate group of ATP to the nucleophilic 3-OH group of shikimate by an associative reaction mechanism (Schlichting & Reinstein, 1997 ▶; Bellinzoni et al., 2006 ▶). Thus, the differences observed in the MtSK–ADP–shikimate ternary complex in the absence of Mg2+ may contribute to understanding of the MtSK chemical mechanism.
3.3. Influence of chloride ion on the structure of MtSK
In our structure of the MtSK–MgADP binary complex, monomer C of the MtSK tetramer does not contain chloride ion. This ion seems to influence the position of the ADP molecule and the position of the shikimate molecule. This monomer presents a conformation that is more open than the other monomers (Fig. 3 ▶). Furthermore, the LID domain of monomer C has a mean B factor that is higher than those of the other monomers (not shown). The ADP molecule bound to monomer C is shifted by approximately 3.0 Å in the active site (Fig. 3 ▶) compared with the other monomers of the MtSK tetramer or with other structures of MtSK in complex with ADP. These results are in agreement with the kinetic and spectroscopic data previously obtained by Cerasoli et al. (2003 ▶). This work shows that the chloride ion increases the stability of the E. chrysanthemi SK structure and that this same ion also influences the affinity of ADP for SK. Thus, the increase in the protein stability may be a consequence of chloride favouring the SK structure in its closed state.
Figure 3.

Superposition of the chains of the structure of the MtSK–MgADP binary complex obtained in the absence of shikimate. (a) Chains A (yellow) and C (light blue), (b) chains B (green) and C (light blue) and (c) chains B (green) and D (red).
The chloride ion bound to the MtSK active site seems to be part of an intricate network formed of water molecules, residues of the LID and SB domains, ADP and shikimate. This network of interactions appears to cause closure of the structure (Fig. 4 ▶). In all structures of MtSK reported so far and in the three monomers of the MtSK tetramer, an interaction network involving three water molecules is formed in the active site (Fig. 4 ▶). These water molecules bridge the interactions between ADP, chloride and magnesium ion and MtSK. In our MtSK–ADP–shikimate ternary complex, water 306 occupies a similar position to the Mg2+ ion. In the absence of chloride ion, this phenomenon does not occur and furthermore the formation of this intricate network that can induce the opening of the structure is avoided.
Figure 4.

Pattern of water bonding in the active site of MtSK. (a) Structure at 1.93 Å resolution (chloride ion and shikimate present, magnesium ion absent), (b) monomer B of the tetramer (chloride and magnesium ions present, shikimate absent), (c) monomer C of the tetramer (magnesium ion present, shikimate and chloride ion absent). The figures were generated using PyMOL (DeLano, 2004 ▶).
3.4. Influence of shikimate in the structure of MtSK
The crystals of the MtSK–MgADP binary complex in the absence of shikimate were obtained in space group P212121, which has not been previously described for MtSK. The asymmetric unit presents four MtSK monomers that form a tetramer. The absence of shikimate from the crystallization conditions influences the crystal packing and also the conformation of the SB and LID domains of MtSK, as observed by Pereira et al. (2004 ▶) and Dhaliwal et al. (2004 ▶). In accordance with this, Gan and coworkers recently solved the apo-MtSK and MtSK–shikimate binary complex structures and suggested that shikimate binding defines the conformational change of the protein that arises when shikimate is bound: the LID domain is ordered and closes over the bound shikimate (Gan et al., 2006 ▶).
4. Conclusion
Here, we report two structures of shikimate kinase from M. tuberculosis: the MtSK–ADP–shikimate and MtSK–MgADP complexes. In the former, we observe the effect of the Mg2+ ion on the structure and in the latter we observe the effect of shikimate on the crystal packing and on the structure of MtSK. The Mg2+ ion seems to influence the position of the hydroxyl groups of the shikimate molecule and some of the residues of the active site of MtSK. The crystal structure of the MtSK–MgADP complex was solved in space group P212121, which has not previously been described for MtSK. In this space group, the MtSK presents a tetramer with 222 symmetry, in which the ribose moiety of the ADP molecule seems to play an important role in the stabilization of the tetramer and the contacts between the monomers, which occur mainly in the LID-domain region. However, one monomer of the tetramer does not contain chloride ion. The absence of this ion seems to cause large changes in the position of the ADP molecule and also causes a large opening of MtSK. This information is accordance with the results obtained by Cerasoli et al. (2003 ▶), which shows the importance of the chloride ion in the stability and the alignment of the substrates in the active site of SK.
We hope that the results described here will shed light on the structural changes of MtSK upon binding of substrate(s) that will be useful for the understanding of the catalytic mechanism and for structure-based drug design of novel inhibitors that may be potential anti-mycobacterial agents.
Supplementary Material
PDB reference: shikimate kinase–ADP–shikimate complex, 2dfn, r2dfnsf
PDB reference: shikimate kinase–MgADP complex, 2dft, r2dftsf
Acknowledgments
This work was supported by grants from FAPESP (SMOLBNet, Proc. 01/07532-0, 03/12472-2, 04/00217-0), CNPq, CAPES and Instituto do Milêncio (CNPq-MCT), DSS, WFA (CNPq, 300851/98-7) and LAB (CNPq, 520182/99-5) are researchers of the Brazilian Council for Scientific and Technological Development.
References
- Bellinzoni, M., Haouz, A., Grana, M., Munier-Lehmann, H., Shepard, W. & Alzari, P. M. (2006). Proteins, 15, 1–5. [DOI] [PMC free article] [PubMed]
- Bentley, R. (1990). Crit. Rev. Biochem. Mol. Biol.25, 307–384. [DOI] [PubMed] [Google Scholar]
- Cerasoli, E., Kelly, S. M., Coggins, J. R., Lapthorn, A. J., Clarke, D. T. & Price, N. C. (2003). Biochim. Biophys. Acta, 1648, 43–54. [DOI] [PubMed] [Google Scholar]
- Coggins, J. R. (1998). In Herbicides and Plant Metabolism, edited by A. Dodge. Cambridge University Press.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Davies, G. M., Barret-Bee, K. J., Jude, D. A., Lehan, M., Nichols, W. W. & Pinder, P. E. (1994). Agents Chemother.38, 403–406. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2004). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Dhaliwal, B., Nichols, C. E., Ren, J., Lockyer, M., Charles, I., Hawhins, A. R. & Stammers, D. K. (2004). FEBS Lett.574, 49–54. [DOI] [PubMed] [Google Scholar]
- Gan, J., Gu, Y., Li, Y., Yan, H. & Ji, X. (2006). Biochemistry, 45, 8539–8545. [DOI] [PubMed] [Google Scholar]
- Gu, Y., Reshetnikova, L., Li, Y., Wu, Y., Yan, H., Singh, S. & Ji, S. (2002). J. Mol. Biol.319, 779–789. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M. & Wüthrich, K. (1996). J. Mol. Graph.14, 51–55. [DOI] [PubMed] [Google Scholar]
- Krell, T., Coggins, J. R. & Lapthorn, A. J. (1998). J. Mol. Biol.278, 983–997. [DOI] [PubMed] [Google Scholar]
- Krell, T., Maclean, J., Boam, D. J., Cooper, A., Resmini, M., Brocklehurst, K., Kelly, S. M., Price, N. C., Lapthorn, A. J. & Coggins, J. (2001). Protein Sci.10, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- McRee, D. E. (1999). J. Struct. Biol.125, 156–165. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Oliveira, J. S., Pinto, C. A., Basso, L. A. & Santos, D. S. (2001). Protein Expr. Purif.22, 430–435. [DOI] [PubMed] [Google Scholar]
- Pereira, J. H., Oliveira, J. S., Canduri, F., Dias, M. V. B., Palma, M. S., Basso, L. A., Santos, D. S. & Azevedo, W. F. Jr (2004). Acta Cryst. D60, 2310–2319. [DOI] [PubMed] [Google Scholar]
- Polikarpov, I., Perles, L. A., de Oliveira, R. T., Oliva, G., Castellano, E. E., Garratt, R. C. & Craievich, A. (1998). Nucl. Instrum. Methods Phys. Res. A, 405, 159–164.
- Ratledge, C. (1982). The Biology of the Mycobacteria, Vol. 1, edited by C. Ratledge & J. L. Stanford, pp. 185–271. London: Academic Press.
- Roberts, F., Roberts, C. W., Johnson, J. J., Kyle, D. E., Krell, T., Coggins, G. H., Milhous, W. K., Tzipoki, S., Ferguson, D. J., Chakrabarti, D. & McLeod, R. (1999). Nature (London), 397, 219–220. [DOI] [PubMed] [Google Scholar]
- Schlichting, I. & Reinstein, J. (1997). Biochemistry, 36, 9290–9296. [DOI] [PubMed] [Google Scholar]
- Uchôa, H. B., Jorge, G. E., Silveira, N. J. F., Câmera, J. C., Canduri, F. & Azevedo, W. F. Jr (2004). Biochem. Biophys. Res. Commun.325, 1481–1486. [DOI] [PubMed] [Google Scholar]
- Vonrhein, C., Schlauderer, G. J. & Schulz, G. F. (1995). Structure, 3, 483–490. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2006). Tuberculosis – Fact Sheet No. 104. http://www.who.int/mediacentre/factsheets.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: shikimate kinase–ADP–shikimate complex, 2dfn, r2dfnsf
PDB reference: shikimate kinase–MgADP complex, 2dft, r2dftsf