

Abstract
The effect of a series of 2-alkylaminoethyl-1,1-bisphosphonic acids against proliferation of the clinically more relevant form of Trypanosoma cruzi, the etiologic agent of American trypanosomiasis (Chagas’ disease), and against tachyzoites of Toxoplasma gondii has been studied. Most of these drugs exhibited an extremely potent inhibitory action against the intracellular form of T. cruzi, exhibiting IC50 values at the low micromolar level. This cellular activity was associated with a strong inhibition of the enzymatic activity of T. cruzi farnesyl diphosphate synthase (TcFPPS), which constitutes a valid target for Chagas’ disease chemotherapy. Compound 17 was an effective agent against amastigotes exhibiting an IC50 value of 0.84 μM, while this compound showed an IC50 value of 0.49 μM against the target enzyme TcFPPS. Interestingly, compound 19 was very effective against both T. cruzi and T. gondii exhibiting IC50 values of 4.1 μM and 2.6 μM, respectively. In this case, 19 inhibited in at least two different enzymes of T. cruzi (TcFPPS and solanesyl diphosphate synthase (TcSPPS); 1.01 μM and 0.25 μM, respectively), while it inhibited TgFPPS in T. gondii. In general, this family of drugs was less effective against the activity of T. cruzi SPPS and against T. gondii growth in vitro. As bisphosphonate-containing compounds are FDA-approved drugs for the treatment of bone resorption disorders, their potential low toxicity makes them good candidates to control tropical diseases.
Introduction
The hemoflagellated protozoan Trypanosoma cruzi is the responsible agent of American trypanosomiasis (Chagas disease), which is the primary source of cardiac death where this disease is endemic1. It has been estimated that over 18 million people are infected and over 40 million individuals are at risk of infection by this parasite.1,2 Of the infected individuals, most of them are in the chronic stage of the disease, in which 30 to 40% will develop serious, often lethal, cardiac and gastrointestinal tract lesions3. T. cruzi is transmitted to humans in the feces of its triatomine insect vector (such as Triatoma infestans or Rhodnius prolixus) and, as other kinetoplastid parasites, it has a complex life cycle, which includes obligatory stages in the mammalian host and in the vector. In the host T. cruzi has two forms: an intracellular dividing form (amastigote), and a non-dividing highly infective bloodstream form (trypomastigote) that can invade host cells. Two forms also occur in the vector: one replicative (epimastigote), and one nonreplicative (metacyclic trypomastigote).4,5
Chemotherapy of Chagas’ disease, which is based on old and fairly non specific drugs empirically discovered, such as nifurtimox (1) and benznidazole (2), remains deficient.6 This illness has also been found in non-endemic countries as a consequence of transfusion of contaminated blood from immigrants.7 For this reason, it is very important to have an efficient agent to eradicate the bloodstream trypomastigotes from blood banks as well. Crystal Violet (3), the only drug employed for blood sterilization and discovered for that purpose some decades ago,8 suffers from some disadvantages, since it was shown to be carcinogenic in in vivo assays (Figure 1).9
Figure 1.
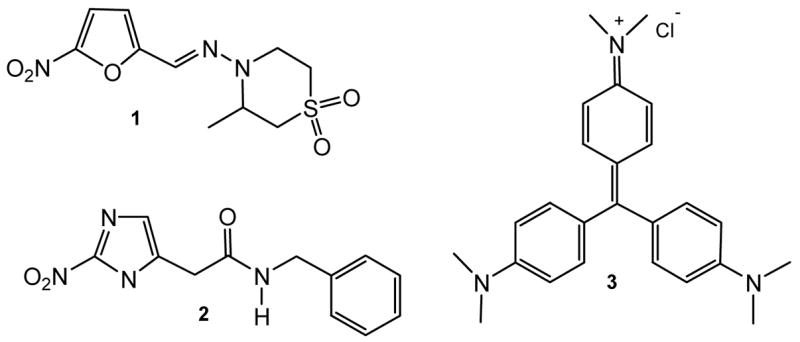
Current Drugs for the Treatment of Chagas’ Disease and Blood Sterilization
Studies of T. cruzi biochemistry and physiology has led to recognize several crucial enzymes for parasite survival as potential targets for the design of new drugs.10 One pathway that has been particularly useful for the identification of new targets against T. cruzi is the isoprenoid pathway. Enzymes studied so far that are involved in the synthesis of sterols11 and farnesyl diphosphate,12 and in protein prenylation13 have been reported to be excellent drug targets against pathogenic parasites. A recent report about the presence of another important prenyltransferase in T. cruzi, a solanesyl diphosphate synthase (TcSPPS), which is involved in the synthesis of ubiquinone, suggested that it is another potential target for chemotherapy.14 For example, farnesyl diphosphate synthase (FPPS) has been demonstrated to be the target of nitrogen-containing- and nitrogen free-bisphosphonates that have activity in vitro and in vivo against trypanosomatids and apicomplexan parasites.6 The crystal structure of TcFPPS at 2 Å resolution has been recently published.15 In addition, the fact that bisphosphonates can target multiple sites of isoprenoid biosynthesis reinforce the idea of testing different enzymes involved in this biosynthetic pathway.16
Several bisphosphonates, which are potent inhibitors of bone resorption, are currently being used for the treatment of several bone disorders such as osteoporosis, Paget’s disease, hypercalcemia, tumor bone metastases, etc.17 Selective action on bone is based on the binding of the bisphosphonate moiety to the bone mineral.17 The selective antiparasitic action of bisphosphonates was proposed to be due to the presence of acidocalcisomes, which, having an equivalent composition to the bone mineral, facilitate their accumulation.18 Representative nitrogen-containing bisphosphonates such as pamidronate (4), alendronate (5), risedronate (6), and ibandronate (7) act by a mechanism that leads to osteoclast apoptosis (Figure 2).19 The target of aminobisphosphonates has been limited to the isoprenoid pathway and more specifically, to protein prenylation.20
Figure 2.
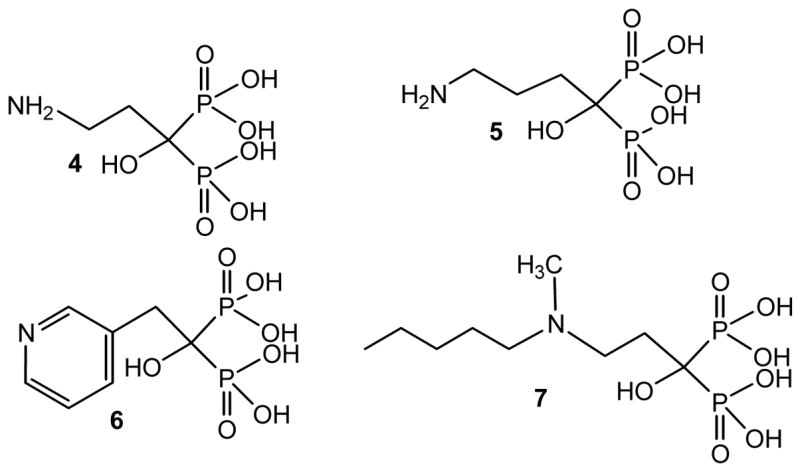
Chemical structure of representative FDA-approved bisphosphonates clinically employed for bone disorders.
Rationale
Nitrogen-containing bisphosphonates were originally found to be effective against T. cruzi in vitro and in vivo without toxicity to the host cells.18 More recently, bisphosphonate derivatives were found to be effective growth inhibitors of pathogenic trypanosomatids other than T. cruzi (T. brucei rhodesiense, Leishmania donovani, and L. mexicana) and apicomplexan parasites (Toxoplasma gondii and Plasmodium falciparum).21–27 In vivo assays of bisphosphonates have shown that risedronate can significantly increase survival of T. cruzi-infected mice.28 The above results indicate that bisphosphonates are promising candidate drugs to treat pathogenic parasitic infections by T. cruzi and other protozoan parasites. In addition, bisphosphonates have the advantage that they are straightforwardly and inexpensively prepared and many compounds bearing the bisphosphonate moiety are FDA-approved drugs for the long-term treatment of several bone disorders. A low toxicity for bisphosphonate-containing drugs might be predictable.
Of special interest are bisphosphonates derived from fatty acids, which were shown to be potent growth inhibitors against the clinically more relevant form of T. cruzi exhibiting IC50 values at the low micromolar range.23–25 Compound 8 arises as main member of this class of bisphosphonates with an IC50 value of 18 μM.23 This cellular activity correlates quite properly with the inhibition of the enzymatic activity towards TcFPPS,24 being a competitive inhibitor with an IC50 value of 1.94 μM and a Ki of 0.40 μM.24 Compound 8 also inhibits the enzymatic activity of T. brucei FPPS,24 and is active in vitro against T. gondii.26 Besides, SAR studies indicated that the replacement of the hydroxyl group at C-1 of 8 by a hydrogen atom to yield 9 resulted in a less effective compound either as inhibitor of the enzymatic activity of TcFPPS or as inhibitor of T. cruzi growth.24 The low efficacy observed for 9, in which the hydroxyl group at C-1 is missing, may be attributable to the lack of ability to coordinate Mg2+ present at the active site of the target enzyme29,30 or due to poor cell permeability. In contrast, the tridentate 8 has three coordination sites versus a bidentate structure that clearly justifies is effectiveness towards TcFPPS. The presence of the amino group would retain the ability to coordinate Mg2+ in a tridentate structure similar to compounds of the type 8. On the other hand, 1-amino-1,1-bisphosphonate derivatives such as 10 were very potent against TcFPPS at the nanomolar range,25 and also were very effective inhibitors of T. cruzi proliferation but to a lesser extent compared with 1-hydroxy-1,1-bisphosphonates.23,25 Moreover, from the X-ray structure of the complex of risedronate with TcFPPS, it can be observed that residue Asp250 forms a hydrogen bond with the hydroxyl group present at the C-1 position of the molecule of risedronate. This observation justifies the poor biological action observed by bisphosphonates where either the hydroxyl group or the amino group is replaced by a hydrogen atom.15 Bearing in mind the above results, it would seem of interest to carry out structural variations on compound 10 taken as lead drug. Therefore, compounds where the relative position of the nitrogen atom respect to the bisphosphonic unit is such as the compound of general formula 11 were envisioned. This family of compounds would retain the ability to coordinate Mg2+ in a tridentate structure. Since compounds 8 and 9 are nitrogen-free bisphosphonates, they could not act as carbocation transition state analogues of the substrate of FPPS as previously postulated.31 In addition, nitrogen-containing bisphosphonates of the class 10 and 11 bear the nitrogen atom in such a position that their protonated form cannot act as an analogue of a transition state of the target enzyme (Figure 3).
Figure 3.
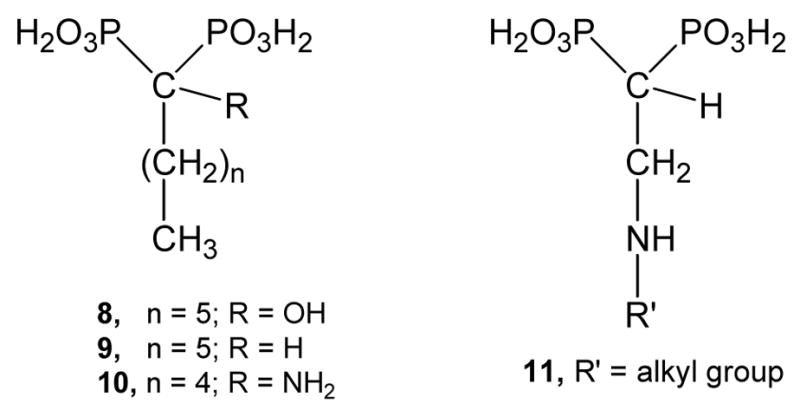
Representative bisphosphonic acids derived from fatty acids.
Results and Discussion
The title compounds were straightforwardly prepared employing tetraethyl ethenylidenbisphosphonate (compound 13) as a Michael acceptor,32 which in turn was easily prepared from tetraethyl methylenebisphosphonate (compound 12) in two steps according to a slightly modified Degenhart protocol.33 Therefore, compound 13 was reacted with the corresponding amine in a Michael-type reaction to afford the respective Michael adduct. Once this synthetic intermediate was at hand, it was hydrolyzed by treatment with concentrated hydrochloric34 acid to afford the title compounds, that is, the corresponding free bisphosphonic acids (14–27). The preparation of this new family of 2-alkylaminoethyl gem-bisphosphonates is illustrated in Scheme 1. Some of these compounds had previously been shown to be herbicidal agents.35,36 Compounds 14–27 were evaluated as growth inhibitors against the amastigote form of T. cruzi, the clinically more relevant form of the parasite. WC-9, a well-known antiparasitic agent, was used as a positive control.37 In addition, the correlation of the cellular activity with the action against its target enzyme (TcFPPS) as well as TcSPPS was studied. Besides, based on previous studies on structurally related bisphosphonates against the opportunistic pathogen T. gondii,26,38 this series of new nitrogen-containing bisphosphonic acids was evaluated against T. gondii and its putative target enzyme TgFPPS.
Scheme 1. Reagents and conditions.
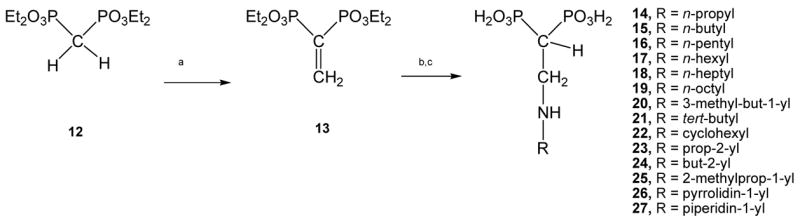
(a) references 40–42; (b) RNH2, CH2Cl2 (anh.), rt, 30 min; (c) (conc.) HCl, reflux, 24h, 94% for 14, 94% for 15, 98% for 16, 85% for 17, 91% for 18, 78% for 19, 88% for 20, 71% for 21, 66% for 22, 79% for 23, 80% for 24, 68% for 25, 77% for 26, 84% for 27.
Most of the title compounds were potent inhibitors of the enzymatic activity of TcFPPS. The efficacy of several compounds on this enzyme qualitatively correlated with their inhibitory action against growth of amastigotes of T. cruzi. Taken compound 17 as an example, this compound was a potent inhibitor of TcFPPS activity with an IC50 in the nanomolar range (0.49 μM). These data correlated completely with the efficacy of this compound as antiparasitic agent. In fact, compound 17 proved to be a potent inhibitor of the amastigote form of the parasite with an IC50 = 0.84 μM. Remarkably, 17 was > 10-fold more potent than WC-9,37,39 a well-known antiparasitic agent employed as positive control, which in turn proved to be more effective than the currently drugs for the treatment of Chagas’ disease such as nifurtimox and benznidazole under the same assays conditions. The cellular activity exhibited by 17 was > 20-fold more potent than 8 (IC50 = 18.1 μM),23 taken as lead drug for the present study. A comparable degree of efficacy as inhibitors of TcFPPS activity was observed for compounds 14 and 18. Surprisingly, in spite of being one order of magnitude more potent than 17 towards TcFPPS, compound 18 exhibited indistinguishable antiproliferative potency against amastigotes compared to WC-9 and a notable less potency than that shown by 17. These compounds have to cross two cell membranes to get to the target enzyme. Compound 18 is apparently less permeable than compound 17 despite its lower charge to mass ratio. For example, compound 14, which was the most effective compound on TcFPPS (IC50 = 38 nM) was devoid of activity against the intracellular form of the parasite. These results were in agreement with our previous results with the isosteric analogue of 14, compound 9, in which the amino group was replaced by a methylene group and which exhibited an IC50 value against TcFPPS of 5.67 μM, and Ki = 0.47 μM and was completely devoid of activity against amastigotes. The above results indicated that the chain length was appropriate in the case of 14 but its permeability should be optimized. Compound 16 was the most potent compound against the intracellular form of T. cruzi (IC50 = 0.54 μM). This activity correlated quite well with the effect of this compound against the target enzyme (IC50 = 1.84 μM). Compound 19 was a very interesting example of a compound effective against growth of both T. cruzi and T. gondii showing IC50 values of 4.1 μM and 2.6 μM, respectively. 19 was very potent against both target enzymes TcFPPS and TcSPPS especially against the latter one with IC50 of 1.01 μM and 0.25 μM, respectively. The inhibitory action towards TcSPPS was not surprising since this compound has the longest side chain among the designed compounds; therefore, it was better recognized by TcSPPS due to substrate similarity. Moreover, 19 also exhibited potent inhibitory action against TgFPPS at the nanomolar range (IC50 = 87 nM). Compounds 17 and 18 were also good inhibitors towards T. cruzi SPPS showing IC50 values of 1.35 μM and 1.69 μM, respectively. However, a high selectivity against TcFPPS, except for 19, was observed in all of the designed compounds. It is interesting to note the lack of trend in inhibition of TcFPPS between compounds 14–19, where there is a logical stepwise variation in structure, but not in activity, and the lack of correlation between growth and enzyme inhibition in other cases (14, 18, 21, 23, 24, 25), suggesting in some cases (16) off-target effects.
Compounds 14 and 17 resulted to be moderately effective against T. gondii with IC50 values of 6.33 μM and 9.37 μM, respectively. The mode of action of these compounds would also be by affecting protein prenylation at the level of TgFPPS (IC50 values of 0.30 μM and 0.14 μM, respectively). Atovaquone was used as a positive control against T. gondii growth (IC50 = 0.29 μM). Recent work40 has indicated that TgFPPS is a bifunctional enzyme, catalyzing the formation of both farnesyl diphosphate (FPP) and geranyl geranyl diphosphate (GGPP). In agreement with that work, we found that the correlation between inhibition of TgFPPS and TcSPPS (a longer chain prenyl synthase) by 12 of the bisphosphonates assayed in this work was high (R2 = 0.69; Figure 4), while there was no correlation between of TgFPPS and TcFPPS inhibition, or between TcFPPS and TcSPPS inhibition (data not shown).
Figure 4. Prenyl synthase inhibition correlation.
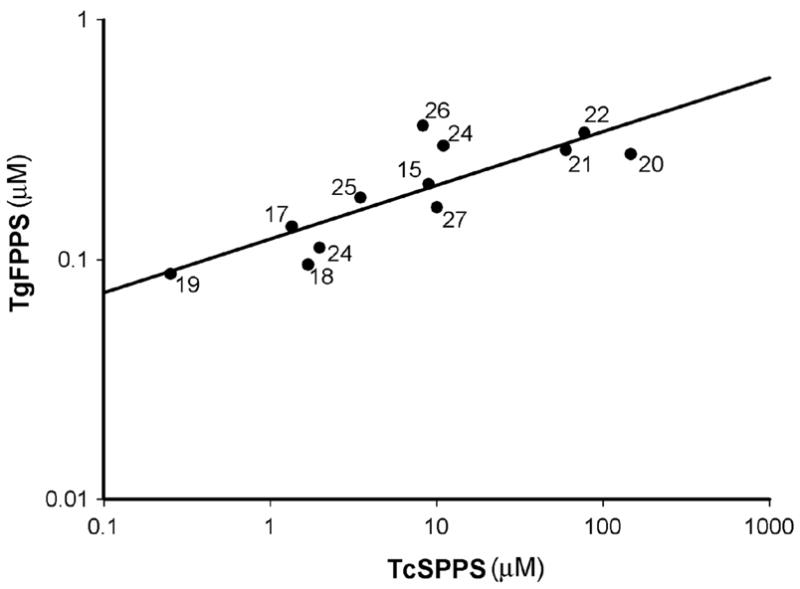
Correlation between TgFPPS and TcSPPS IC50 values, for the bisphosphonates indicated with numbers.
It can be concluded that the isosteric replacement of the methylene group at the C-3 position of 1-alkyl-1,1-bisphosphonates such as compound 9 to give compounds of general formula 11 was of the great effectiveness against both T. cruzi and the target enzyme TcFPPS. Most of the designed compounds were even much more potent than the gem-bisphosphonic acid 8, taken as a reference structure, against both T. cruzi and the target enzyme, T. cruzi farnesyl diphosphate synthase. Most of these compounds proved to be inhibitors of the enzymatic activity of TcSPPS but to a lesser extent than TcFPPS as previously discussed. Moreover, some of these 1-[2-(alkylamino)ethyl] bisphosphonic acids or 1-(3-azaalkyl)-1,1 bisphosphonic acids were shown to be moderately effective anti-T. gondii agents indicating the broad scope that this type of compounds has. Work aimed at optimizing the chemical structure of 1-(3-azaalkyl)-1,1 bisphosphonic acids such as compound 17 and other closely related analogues is currently being pursued in our laboratory.
Experimental Section
General
The glassware used in air- and/or moisture-sensitive reactions was flame-dried and reactions were carried out under dry argon. Unless otherwise noted, chemicals were commercially available and used without further purification. Solvents were distilled before use. Dichloromethane was distilled from phosphorus pentoxide. Nuclear magnetic resonance spectra were recorded with a Bruker AM-500 MHz spectrometer. The 1H NMR spectra are referenced with respect to the residual CHCl3 proton of the solvent CDCl3 at δ = 7.26 ppm. Coupling constants are reported in Hz. 13C NMR spectra were fully decoupled and are referenced to the middle peak of the solvent CDCl3 at δ = 77.0 ppm. 31P NMR spectra are referenced with respect to the peak of 85% H3PO4 as external reference. Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quadruplet; dd, double doublet, etc. Melting points were determined with a Fisher–Johns apparatus and are uncorrected. IR spectra were recorded with a Nicolet Magna 550 spectrometer. Analytical TLC was performed on commercial 0.2 mm aluminum-coated silica gel plates (F254) and visualized by 254 nm UV or immersion in an aqueous solution of (NH4)6Mo7O24 · 4H2O (0.04 M), Ce(SO4)2 (0.003 M) in concentrated H2SO4 (10%). Elemental analyses were conducted by Atlantic Microlab Inc., Norcross, Georgia. The results were within ±0.4% of the theoretical values.
Synthesis of 1-[2-alkylaminoethyl]-1,1-bisphosphonic acids
General Procedure
A solution of compound 13 (10 mmol) in anhydrous methylene chloride (10 mL) was treated with the corresponding amine (10 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 30 min. The solvent was evaporated and the residue was treated with a concentrated aqueous solution of hydrochloric acid (2 mL). The resulting mixture was refluxed for 24 h. The solvent was evaporated and the residue was crystallized from ethanol.
1-[(Prop-1-ylamino)ethyl] 1,1-bisphosphonic Acid (14)
94% Yield; mp 195 °C; 1H NMR (D2O) δ 0.86 (t, J = 7.4 Hz, 3H, H-6); 1.60 (sex, J = 7.2 Hz, 2H, H-5); 2.43 (tt, J = 21.3, 7.0 Hz, 2 H, H-1), 2.95 (t, J = 7.1 Hz, 2H, H-4), 3.33 (dt, J = 14.0, 7.1 Hz, 2H, H-2); 13C NMR (D2O) δ 10.04 (C-6), 19.07 (C-5), 35.98 (t, J = 121.7 Hz, C-1), 44.58 (C-2), 49.17 (C-4); 31P NMR (D2O) δ 16.14. Anal. (C5H15NO6P2) Calcd. C 24.30, H 6.12, N 5.67. Found C 24.01, H 6.12, N 5.70.
1-[(n-But-1-ylamino)ethyl] 1,1-bisphosphonic Acid (15)
92% Yield; mp 214 °C; IR (BrK, cm−1) 3101, 2314, 1273, 1180, 956; 1H NMR (D2O) δ 0.83 (t, J = 7.4 Hz, 3H, H-7), 1.30 (sex, J = 7.5, 2H, H-6), 1.58 (pent, J = 7.5 Hz, 2H, H-5), 2.42 (tt, J = 21.7, 7.0 Hz, 1H, H-1), 3.01 (t, J = 7.4 Hz, 2H, H-4), 3.35 (dt, J = 14.3, 7.3 Hz, 2H, H-2); 13C NMR (D2O) δ 12.6 (C-7), 20.0 (C-6), 27.5 (C-5), 36.1 (t, J = 120.3 Hz, C-1), 44.7 (C-2), 47.4 (C-4); 31P NMR (D2O) δ 15.99. Anal. (C6H17NO6P2) Calcd. C 27.59, H 6.56, N 5.36. Found C 27.94, H 6.72, N 5.57.
1-[(n-Pent-1-ylamino)ethyl] 1,1-bisphosphonic Acid (16)
98% Yield; mp 204 °C; IR (BrK, cm−1) 3101, 2310, 1273, 1180, 960; 1H NMR (D2O) δ 0.78 (t, J = 6.9 Hz, 3H, H-8), 1.26 (m, 4H, H-6, H-7), 1.60 (q, J = 7.3 Hz, 2H, H-4), 1.59 (q, J = 7.4 Hz, 2H, H-4), 2.41 (tt, J = 21.4, 7.3 Hz, 1H, H-1), 3.01 (t, J = 7.3 Hz, 2H, H-4), 3.35 (dt, J = 14.3, 7.2 Hz, 2H, H-2); 13C NMR (D2O) δ 13.0 (C-8), 21.4 (C-7), 25.1 (C-6), 27.7 (C-5), 36.1 (t, J = 121.2 Hz, C-1), 44.7 (C-2), 47.6 (C-4); 31P NMR (D2O) δ15.99. Anal. (C7H19NO6P2· · ½H2O) Calcd. C 29.59, H 7.09, N 4.93. Found C 29.78, H 6.86, N 4.93.
1-[(n-Hex-1-ylamino)ethyl] 1,1-bisphosphonic Acid (17)
85% Yield; mp 212 °C; IR (BrK, cm−1) 3105, 2307, 1712, 1273, 1176, 956; 1H NMR (D2O) δ 0.76 (t, J = 6.6 Hz, 3H, H-9), 1.21 (m, 4H, H-7, H-8), 1.28 (m, 2H, H-6), 1.59 (pent, J = 7.4 Hz, 2H, H-5), 2.42 (tt, J = 21.5, 7.2 Hz, 1H, H-1), 3.00 (t, J = 7.4 Hz, 2H, H-4), 3.34 (dt, J = 14.3, 7.3 Hz, 2H, H-2); 13C NMR (D2O) δ 13.1 (C-9), 21.6 (C-8), 25.2 (C-7), *25.4 (C-6),* 30.3 (C-5), 36.1 (t, J = 120.7 Hz, C-1), 44.7 (C-2), 47.7 (C-4); 31P NMR (D2O) δ 16.02. Anal. (C8H21NO6P2) Calcd. C 33.22, H 7.32, N 4.84. Found C 33.39, H 7.13, N 4.95.
1-[(n-Hept-1-ylamino)ethyl] 1,1-bisphosphonic Acid (18)
91% Yield; mp 207 °C; IR (BrK, cm−1) 3094, 2310, 1269, 1180, 1003; 1H NMR (D2O) δ 0.76 (t, J = 6.0 Hz, 3H, H-9), 1.18–1.24 (m, 8H, H-8 H-7 H-6 H-5), 1.59 (q, J = 7.1 Hz, 2H, H-4), 2.43 (tt, J = 21.4, J = 7.0 Hz, 1H, H-1), 3.00 (t, J = 6.9 Hz, 2H, H-3), 3.34 (dt, J = 13.9, 7.1 Hz, 2H, H-2); 13C NMR (D2O) δ 13.3 (C-9), 21.8 (C-8), 25.4 (C-7),* 25.5 (C-6),* 27.8 (C-4), 30.7 (C-5), 36.0 (t, J = 120.3 Hz, C-1), 44.7 (C-2), 47.7 (C-3); 31P NMR (D2O) δ 15.61. Anal. (C9H13NO6P2) Calcd. C 35.65, H 7.65, N 4.62. Found C 35.38, H 7.59, N 4.80.
1-[(n-Oct-1-ylamino)ethyl] 1,1-bisphosphonic Acid (19)
78% Yield; mp 202 °C; 1H NMR (D2O) δ 0.70 (t, J = 6.9 Hz, 3H, H-11), 1.13 (m, 10H, H-6, H-7, H-8, H-9, H-10), 1.36 (pent, J = 7.4 Hz, 2H, H-5), 1.73 (tt, J = 21.0, 6.9 Hz, 1H, H-1), 2.48 (t, J = 7.6 Hz, 2H, H-4), 2.84 (dt, J = 14.3, 6.2 Hz, 2H, H-2);. 13C NMR (D2O) δ 13.3 (C-11), 21.9 (C-10), 26.5 (C-9), 28.1 (C-8), 28.3 (C-7), 28.5 (C-6), 31.1 (C-5), 40.6 (t, J = 116.7 Hz, C-1), 48.1 (C-2), 48.3 (C-4); 31P NMR (D2O) δ 17.99. Anal. (C10H25NO6P2) Calcd. C 37.86, H 7.94, N 4.41. Found C 37.69, H 7.85, N 4.40.
1-[(3-Methyl-but-1-ylamino)ethyl] 1,1-bisphosphonic Acid (20)
88% Yield; mp 231 °C; 1H NMR (D2O) δ 0.83 (d, J = 6.4 Hz, 6 H, H-6), 1.49 (dd, J = 15.1, 7.1 Hz, 2 H, H-4), 1.57 (hept, J = 6.6 Hz, H, H-5), 2.40 (tt, J = 21.5, 7.3 Hz, 1 H, H-1), 3.04 (t, J = 7.7 Hz, 2 H, H-3), 3.35 (dt, J = 14.3, 7.2 Hz, 2 H, H-2); 13C NMR (D2O) δ 21.29 (C-6), 25.03 (C-5), 34.20 (C-4), 36.18 (t, J = 120.3 Hz, C-1), 44.85 (C-2), 46.09 (C-3); 31P NMR (D2O) δ 15.87. Anal. (C7H19NO6P2)) Calcd. C 30.55, H 6.96, N 5.09. Found C 30.79, H 6.96, N 5.08.
1-[(tert-Butylamino)ethyl] 1,1-bisphosphonic Acid (21)
71% Yield; mp 238 °C; 1H NMR (D2O) δ 1.28 (s, 9 H, C(CH3)3), 2.31 (tt, J = 21.4, 7.3 Hz, 1 H, H-1), 3.31 (dt, J = 14.2, 7.3 Hz, 2 H, H-2); 13C NMR (D2O) δ 24.98 C(CH3)3), 36.34 (t, J = 120.8 Hz, C-1), 38.86 (C-2), 57.05 (C-3); 31P NMR (D2O) δ 16.04. Anal. (C6H17NO6P2) Calcd. C 27.59, H 6.56, N 5.36. Found C 27.21, H 6.55, N 5.14.
1-[(Cyclohexylamino)ethyl] 1,1-bisphosphonic Acid (22)
66% Yield; mp 227 °C; 1H NMR (D2O) δ 2.38 (tt, J = 21.5, 7.3 Hz, 3.07 (m, 1 H, H-), 3.35 (dt, J = 14.3, 7.2 Hz, 2 H, H-); 13C NMR (D2O) δ 23.64, 24.42, 28.92, 36.01 (t, J = 121.7 Hz, C-1), 41.76, 57.09; 31P NMR (D2O) δ 16.12. Anal. (C8H19NO6P2) Calcd. C 33.46, H 6.67, N 4.88. Found C 33.38, H 6.64, N 5.01.
1-[(Prop-2-ylamino)ethyl] 1,1-bisphosphonic Acid (23)
79% Yield; mp 227–228 °C; 1H NMR (D2O) δ 1.23 (d, J = 6.6 Hz, 6H, H-5), 2.37 (tt, J = 21.5, 7.2 Hz, 1H, H-1), 3.33 (dt, J = 14.2, 7.1 Hz, 2H, H-2), 3.37 (sept, J = 6.5 Hz, 1H, H-4); 13C NMR (D2O) δ 18.3 (C-5), 36.1 (t, J = 121.7 Hz, C-1), 42.0 (C-2), 50.8 (C-4); 31P NMR (D2O) δ 16.04. Anal. (C5H15NO6P2)) Calcd. C 24.30, H 6.12, N 5.67. Found C 24.50, H 6.27, N 5.74.
1-[(But-2-ylamino)ethyl] 1,1-bisphosphonic Acid (24)
80% Yield; mp 197 °C; 1H NMR (D2O) δ 0.83 (dist. t, J = 7.4 Hz, 3H, H-6), 1.16 (d, J = 6.6 Hz, 3H, CH3 at C-4), 1.49 (m, 1H, H-5a), 1.60 (m, 1H, H-5b), 2.38 (tt, J = 21.7, 7.3 Hz, 1H, H-1), 3.15 (m, 1H, H-4), 3.26 (m, 1H, H-2a) 3.37 (m, 1H, H-2b); 13C NMR (D2O) δ 8.7 (C-6), 15.1 (CH3 at C-4), 35.8 (t, J = 122.1 Hz, C-1), 25.9 (C-5), 41.9 (C-2), 56.0 (C-4); 31P NMR (D2O) δ 16.14 (d, J = 7.4 Hz, Pa), 16.14 (d, J = 7.4 Hz, Pb). Anal. (C6H17NO6P2)) Calcd. C 27.60, H 6.56, N 5.36. Found C 27.81, H 6.65, N 5.44.
1-[(2-Methylprop-1-ylamino)ethyl] 1,1-bisphosphonic Acid (25)
68% Yield; mp 220–222 °C; 1H NMR (D2O) δ 0.89 (d, J = 6.9 Hz, 6H, H-6), 1.90 (n, J = 6.8 Hz, 1H, H-5), 2.42 (tt, J = 21.3, 7.3 Hz, 1H, H-1), 2.84 (d, J = 7.0 Hz, 1H, H-4), 3.34 (dt, J = 14.0, 7.4 Hz, 2H, H-2); 13C NMR (D2O) δ 18.9 (C-6), 25.5 (C-5), 35.9 (t, J = 121.2 Hz, C-1), 45.1 (C-2), 54.6 (C-4); 31P NMR (D2O) δ 16.36. Anal. (C6H17NO6P2) Calcd. C 27.60, H 6.56, N 5.36. Found C 27.31, H 6.50, N 5.20.
1-[(2-Pyrrolidin-1-ylamino)ethyl] 1,1-bisphosphonic Acid (26)
77% Yield; mp 235–236 °C; 1H NMR (D2O) δ 1.95 (m, 2H, H-), 2.07 (m, 2H, H-), 2.53 (tt, J = 21.6, 7.7 Hz, 1H, H-1), 3.05 (2H, H-), 3.48 (ddd, J = 14.8, 12.6, 7.8 Hz, 2H, H-), 3.65 (p, J = 5.6 Hz, 2H, H-); 13C NMR (D2O) δ 22.8 (C-5), 36.1 (t, J = 121.0 Hz, C-1), 52.8 (C-2), 54.4 (C-4); 31P NMR (D2O) δ 15.51. Anal. (C6H15NO6P2 • 2H2O) Calcd. C 24.42, H 6.49, N 4.75. Found C 24.70, H 6.47, N 4.66.
1-[(2-Piperidin-1-ylamino)ethyl] 1,1-bisphosphonic Acid (27)
84% Yield; mp 226–228 °C; 1H NMR (D2O) δ 1.38 (m, 1H, H-), 1.58 (m, 2H, H-), 1.69 (m, 1H, H-), 1.85 (m, 2H, H-), 2.61 (tt, J = 21.4, 7.7 Hz, 1H, H-1), 2.86 (t, J = 11.1 Hz, 2H, H-), 3.31 (m, 2H, H-), 3.49 (t, J = 11.1 Hz, 2H, H-); 13C NMR (D2O) δ 21.3 (C-5), 23.4 (C-5), 34.0 (t, J = 121.0 Hz, C-1), 53.4 (C-2), 54.3 (C-4); 31P NMR (D2O) δ15.60. Anal. (C7H17NO6P2) Calcd. C 30.78, H 6.27, N 5.13. Found C 30.33, H 6.30, N 4.99.
Drug Screening
Experiments on the intracellular form of the parasite were conducted on T. cruzi-infected L6E9 myoblasts (Y strain) as described before.41,42 Gamma-irradiated L6E9 myoblasts (1 × 107 cells/plate) in DMEM medium containing 20% FCS were plated in 12-well tissue culture plates and incubated at 37 °C in a 5% CO2 atmosphere for 24 h. After 24 h, wells were washed once and fresh media was added containing 4.17 × 106 trypomastigotes or amastigotes/well in DMEM medium. One well was left without parasites for control. After 2 h of incubation at 37 °C in a 7% CO2 atmosphere, cultures were washed twice with Hank’s solution, and culture medium was replaced to remove extracellular parasites. At this time, 0.5–1 μCi of [5,6–3H]uracil/well (specific activity, 40–50 Ci/mmol) and drug solutions in water were added. Two wells were left for infection control. Cultures were incubated for 72 h. Incorporation of the [5,6-3H]uracil into trichloroacetic acid (TCA)-precipitable material was measured at day 3. The supernatants from the monolayers were transferred to glass tubes, the cells were dissolved with 1.3 ml of 1% sodium dodecyl sulfate containing 100 μg of cold uracil per ml, and the suspension was transferred to the glass tubes. The wells were rinsed with 3 ml of 5 % TCA (ice-cold) which was combined with the previous suspension. The samples were maintained in ice for 15 min and collected on glass fiber filters (Whatman GF/B) by using a sampling manifold (Millipore, Bedford, MA). After filtering, the tubes were rinsed twice with 4 ml of 5% TCA and the filters were rinsed twice with TCA and once with 95% ethanol. After drying the filters, they were placed in scintillation vials containing 4–5 ml of scintillation cocktail Ecolume. Vials were vortexed for 20 seconds and counted. The percent inhibition was calculated by employing the following formula: Percent inhibition = 100 − (ΔCd × 100)/ΔCc, where ΔCc and ΔCd are the differences in the counts per minute of control cultures and drug-treated cultures, respectively.
Experiments on T. gondii tachyzoites were carried out as previously published.43 Briefly, T.gondii tachyzoites of the m2m3 clone expressing yellow fluorescence protein43 were routinely maintained in vitro in human foreskin fibroblast monolayers (HFF) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 mM pyruvate, at 37 °C in a humid 5% CO2 atmosphere. Confluent HFF monolayers grown in 96-well black plates with optical bottoms (Falcon/Becton-Dickinson, Franklin Lakes, NJ) were used and drugs dissolved in the same medium and serially diluted in the plates. Freshly isolated tachyzoites were filtered through a 3 μm filter and passed through a 22 gauge needle, before use. The cultures were inoculated with 104 tachyzoites/mL in the same media. The plates were incubated at 37 °C and read daily in a Molecular Devices fluorescent plate reader. To preserve sterility the plates were read with covered lids, and both excitation (510 nm) and emission (540 nm) were read from the bottom.41 For the calculation of the IC50, the percent of growth inhibition was plotted as a function of drug concentration by fitting the values to the function: I = Imax C/(IC50 + C), where I is the percent inhibition, Imax = 100% inhibition, C is the concentration of the inhibitor, and IC50 is the concentration for 50% growth inhibition.
TcFPPS and TgFPPS Assays and Product Analysis
For TcFPPS31,44,45 100 μL of assay buffer (10 mM Hepes, pH 7.4, 5 mM MgCl2, 2 mM dithiothreitol, 100μM [4-14C]IPP (10 μCi/μmol)), and 100 μM DMAPP were prewarmed to 37 °C. The assay was initiated by the addition of recombinant protein (10–20 ng). The assay was allowed to proceed for 30 min at 37 °C and was quenched by the addition of 6 M HCl (10 μL). The reactions were made alkaline with 6.0 M NaOH (15 μL), diluted in water (0.7 mL), and extracted with of hexane (1 mL). The hexane solution was washed with water and transferred to a scintillation vial for counting. One unit of enzyme activity was defined as the activity required to incorporate 1 nmol of [4-14C]IPP into [14-14C]FPP in 1 min. For TgFPPS the assay conditions were as described above except that the buffer contained 1 mM MgCl2.
TcSPPS assay
The activity of the enzyme was determined by a radiometric assay based on that described before.14,46 Briefly, 100 μl of assay buffer (100 mM Tris-HCl buffer, pH 7.4, 1 mM MgCl2, 1% (v/v) Triton X-100, 100μM [4-14C]IPP (10μCi/μmol)), and 50μM GGPP was prewarmed to 37 °C. The assay was initiated by the addition of 10–20 ng of recombinant protein. The assay was allowed proceed for 30 min at 37 °C and was quenched by chilling quickly in an ice bath. The reaction products were extracted with 1 ml of 1-butanol saturated with water. The organic layer was washed with water saturated with NaCl, and transferred to a scintillation vial with 4 ml of scintillation solution Ecolume for counting. One unit of enzyme activity was defined as the activity required to incorporate 1 nmol of [4-14C]IPP into [4-14C]FPP in 1 min.
Supplementary Material
Table 1.
| Comp. | IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) |
|---|---|---|---|---|---|
| T. cruzi amastigotes | T. gondii tachyzoites | TcFPPS | TcSPPS | TgFPPS | |
| 14 | > 50 | 6.33 | 0.038 ± 0.008 | 11.01 ± 1.87 | 0.298 ± 0.035 |
| 15 | 4.8 | 48.67 | 2.28 ± 0.34 | 8.96 ± 2.18 | 0.206 ± 0.010 |
| 16 | 0.54 | >50 | 1.84 ± 0.25 | 16.79 ± 4.37 | 0.080 ± 0.023 |
| 17 | 0.84 | 9.37 | 0.49 ± 0.12 | 1.35 ± 0.22 | 0.137 ± 0.087 |
| 18 | 10.0 | >50 | 0.058 ± 0.009 | 1.69 ± 0.39 | 0.095 ± 0.024 |
| 19 | 4.1 | 2.60 | 1.014 ± 0.457 | 0.252 ± 0.050 | 0.087 ± 0.038 |
| 20 | 10.0 | 36.94 | 0.42 ± 0.18 | 147.1 ± 77.2 | 0.275 ± 0.041 |
| 21 | 10.0 | 42.84 | 1.21 ± 0.13 | 59.8 ± 13.9 | 0.286 ± 0.059 |
| 22 | 36.3% at 10 μM | 57.3 | 0.013 ± 0.003 | 77.4 ± 4.09 | 0.337 ± 0.138 |
| 23 | > 50 | 69.81 | 1.399 ± 0.496 | 0.761 ± 0.471 | 0.253 ± 0.069 |
| 24 | > 50 | > 100 | 0.944 ± 0.211 | 1.980 ± 0.359 | 0.112 ± 0.004 |
| 25 | > 50 | 74.47 | 0.515 ± 0.104 | 3.493 ± 2.678 | 0.181 ± 0.050 |
| 26 | 14 | 13.30 | 0.760 ± 0.316 | 8.276 ± 6.474 | 0.361 ± 0.203 |
| 27 | 6.1 | 70.60 | 0.287 ± 0.021 | 10.034 ± 2.307 | 0.165 ± 0.045 |
| WC-9 | 12.0 |
Acknowledgments
This work was supported by grants from the National Research Council of Argentina (PIP 5508), ANPCyT (PICT2004 #21897), and the Universidad de Buenos Aires (X-252) to J.B.R., and the U.S. National Institutes of Health to R. D. (AI-68647) and S. N. J. M. (AI68467). G. G. L. thanks the Ellison Medical Foundation for a Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Urbina JA, Docampo R. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Moncayo A. Eleventh Programme Report of the UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases (TDR) World Health Organization; Geneva: 1995. pp. 67–75. [Google Scholar]
- 3.Docampo R. Curr Pharm Des. 2001;7:1157–1164. doi: 10.2174/1381612013397546. [DOI] [PubMed] [Google Scholar]
- 4.Brener Z. Annu Rev Microbiol. 1973;27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 5.De Souza W. Int Rev Cytol. 1984;86:197–283. doi: 10.1016/s0074-7696(08)60180-1. [DOI] [PubMed] [Google Scholar]
- 6.García Liñares G, Ravaschino EL, Rodriguez JB. Curr Med Chem. 2006;13:335–360. doi: 10.2174/092986706775476043. [DOI] [PubMed] [Google Scholar]
- 7.Galel SA, Kirchhoff LV. Transfusion. 1996;36:227–231. doi: 10.1046/j.1537-2995.1996.36396182140.x. [DOI] [PubMed] [Google Scholar]
- 8.Nussenzweig V, Sonntag R, Biancalana A, Pedreira de Fleitas JL, Amato Neto V, Kloetzel J. Hospital (Rio de Janeiro) 1953;44:731–744. [PubMed] [Google Scholar]
- 9.Docampo R, Moreno SNJ. Rev Biochem Toxicol. 1985;7:159–204. [Google Scholar]
- 10.De Castro SL. Acta Tropica. 1993;53:83–98. doi: 10.1016/0001-706x(93)90021-3. [DOI] [PubMed] [Google Scholar]
- 11.Urbina JA. Curr Pharm Des. 2002;8:287–295. doi: 10.2174/1381612023396177. [DOI] [PubMed] [Google Scholar]
- 12.Docampo R, Moreno SNJ. Curr Drug Targets Infect Disord. 2001;1:51–61. doi: 10.2174/1568005013343191. [DOI] [PubMed] [Google Scholar]
- 13.Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. Mol Biochem Parasitol. 2003;126:155–163. doi: 10.1016/s0166-6851(02)00282-7. [DOI] [PubMed] [Google Scholar]
- 14.Ferella M, Montalvetti A, Rohloff P, Miranda K, Fang J, Reina S, Kawamukai M, Búa J, Nilsson D, Pravia C, Katzin A, Cassera MB, Åslund L, Andersson B, Docampo R, Bontempi EJ. J Biol Chem. 2006;281:39339–39348. doi: 10.1074/jbc.M607451200. [DOI] [PubMed] [Google Scholar]
- 15.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel LM. Proteins. 2006;62:80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 16.Guo R–T, Cao R, Liang P–H, Ko T–P, Chang T–H, Hudock MP, Jeng W–Y, Chen CK–M, Zhang Y, Song Y, Kuo C–J, Yin F, Oldfield E, Wang AH–J. Proc Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reszka AA, Rodan GA. Mini Rev Med Chem. 2004;4:711–719. [PubMed] [Google Scholar]
- 18.Urbina JA, Moreno B, Vierkotter S, Oldfield E, Payares G, Sanoja C, Bailey BN, Yan W, Scott DA, Moreno SNJ, Docampo R. J Biol Chem. 1999;274:33609–33615. doi: 10.1074/jbc.274.47.33609. [DOI] [PubMed] [Google Scholar]
- 19.Hughes DE, Wright KR, Uy HL, Sasaki A, Yoneda T, Roodman GD, Mundy GR, Boyce BF. J Bone Miner Res. 1995;10:1478–1487. doi: 10.1002/jbmr.5650101008. [DOI] [PubMed] [Google Scholar]
- 20.Rogers MJ, Frith JC, Luckman SP, Coxon FP, Benford HL, Monkkonen J, Auriola S, Chilton KM, Russell RG. Bone. 1999;24:73S–79S. doi: 10.1016/s8756-3282(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 21.Martin MB, Sanders JM, Kendrick H, De Luca-Fradley K, Lewis JC, Grimley JS, Van Brussel EM, Olsen JR, Meints GA, Burzynska A, Kafarski P, Croft SL, Oldfield E. J Med Chem. 2002;45:2904–2914. doi: 10.1021/jm0102809. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S, Chan JM, Lea CR, Meints GA, Lewis JC, Tovian ZS, Flessner RM, Loftus TC, Bruchhaus I, Kendrick H, Croft SL, Kemp RG, Kobayashi S, Nozaki T, Oldfield E. J Med Chem. 2004;47:175–187. doi: 10.1021/jm030084x. [DOI] [PubMed] [Google Scholar]
- 23.Szajnman SH, Bailey BN, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2001;11:789–792. doi: 10.1016/s0960-894x(01)00057-9. [DOI] [PubMed] [Google Scholar]
- 24.Szajnman SH, Montalvetti A, Wang Y, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2003;13:3231–3235. doi: 10.1016/s0960-894x(03)00663-2. [DOI] [PubMed] [Google Scholar]
- 25.Szajnman SH, Ravaschino EL, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2005;15:4685–4690. doi: 10.1016/j.bmcl.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 26.Ling Y, Sahota G, Odeh S, Chan JMW, Araujo FG, Moreno SNJ, Oldfield E. J Med Chem. 2005;48:3130–3140. doi: 10.1021/jm040132t. [DOI] [PubMed] [Google Scholar]
- 27.Martin MB, Grimley JS, Lewis JC, Heath HT, 3rd, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SN, Docampo R, Croft SL, Oldfield E. J Med Chem. 2001;44:909–916. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 28.Bouzahzah B, Jelicks LA, Morris SA, Weiss LM, Tanowitz HB. Parasitol Res. 2005;96:184–187. doi: 10.1007/s00436-005-1331-9. [DOI] [PubMed] [Google Scholar]
- 29.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J Biol Chem. 2003;278:18401–18407. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 30.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Proc Natl Acad Sci USA. 1996;93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R. J Biol Chem. 2001;276:33930–33937. doi: 10.1074/jbc.M103950200. [DOI] [PubMed] [Google Scholar]
- 32.Szajnman SH, García Liñares GE, Moro P, Rodríguez JB. Eur J Org Chem. 2005:3687–3696. [Google Scholar]
- 33.Degenhardt CR, Burdsall DC. J Org Chem. 1986;51:3488–3490. [Google Scholar]
- 34.Wasielewski C, Antczak K. Synthesis. 1981:540–541. [Google Scholar]
- 35.Cromartie TH, Fisher K. J PCT Int Appl. 1995 WO 95/34207 A1. [Google Scholar]
- 36.Fisher KJ, Woolard FX, Leadbetter MR, Gerdes JM. 5,728,650 US Patent. 1998
- 37.Cinque GM, Szajnman SH, Zhong L, Docampo R, Rodriguez JB, Gros EG. J Med Chem. 1998;41:1540–1554. doi: 10.1021/jm970860z. [DOI] [PubMed] [Google Scholar]
- 38.Rodrigues CO, Scott DA, Bailey B, de Souza W, Benchimol M, Moreno B, Urbina JA, Oldfield E, Moreno SNJ. Biochem J. 2000;349:737–745. doi: 10.1042/bj3490737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urbina JA, Concepcion JL, Montalvetti A, Rodriguez JB, Docampo R. Antimicrob Agents Chemother. 2003;47:2047–2050. doi: 10.1128/AAC.47.6.2047-2050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ling Y, Li Z–H, Miranda K, Oldfield E, Moreno SNJ. J Biol Chem. 2007;282:30804–30816. doi: 10.1074/jbc.M703178200. [DOI] [PubMed] [Google Scholar]
- 41.Ravaschino EL, Docampo R, Rodriguez JB. J Med Chem. 2006;49:426–435. doi: 10.1021/jm050922i. [DOI] [PubMed] [Google Scholar]
- 42.Yan W, Moreno SN. J Immunol Methods. 1998;220:123–128. doi: 10.1016/s0022-1759(98)00155-0. [DOI] [PubMed] [Google Scholar]
- 43.Gubbels M–J, Li C, Striepen B. Antimicrob Agents Chemother. 2003;47:309–316. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montalvetti A, Fernandez A, Sanders JM, Ghosh S, Van Brussel E, Oldfield E, Docampo R. J Biol Chem. 2003;278:17075–17083. doi: 10.1074/jbc.M210467200. [DOI] [PubMed] [Google Scholar]
- 45.Ogura K, Nishino T, Shinka T, Seto S. Methods Enzymol. 1985;110:167–171. [Google Scholar]
- 46.Rilling HC. Eukaryotic prenyltransferases. Methods Enzymol. 1985;110:145–152. doi: 10.1016/s0076-6879(85)10069-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.