

The crystal structure of the sweet protein MNEI at 1.15 Å resolution reveals networks of alternate conformations and stably bound negative ions.
Keywords: monellin, sweet protein, sweet taste, T1R2–T1R3 receptor
Abstract
The X-ray crystal structure of a single-chain monellin protein (MNEI) has been determined at 1.15 Å resolution. The model was refined to convergence employing anisotropic displacement parameters and riding H atoms to produce a final model with R work and R free values of 0.132 and 0.162, respectively. The crystal contains a single MNEI protein in the asymmetric unit and unusually lacks the dimer interface observed in all previous crystal structures of monellin and its single-chain derivatives. The high resolution allowed a more detailed view of MNEI than previously possible, with 38 of the 96 residues modelled with alternative side-chain conformations, including four core residues Thr12, Cys41, Leu62 and Ile75. Four stably bound negative ions were also located, providing new insight into potential electrostatic interactions of MNEI with the largely negatively charged surface of the sweet taste receptor T1R2–T1R3.
1. Introduction
The protein monellin is a highly potent sweetener: on a molar basis, it is many thousands of times sweeter than sucrose (Morris & Cagan, 1972 ▶). Isolated from the berries of the African plant Dioscoreophyllum cumminsii (Morris & Cagan, 1972 ▶), monellin is perceived as sweet by humans and some Old World primates, but is not preferred by other mammals (Hellekant et al., 1976 ▶, 1993 ▶). Like other sweet proteins (e.g. brazzein, thaumatin), monellin is of potential use as a noncarbohydrate sweetener and could be particularly beneficial to individuals such as diabetics who must control sugar intake. In its natural form, monellin is a 10.7 kDa protein composed of two chains (A and B, of 44 and 50 amino acids, respectively) and is unstable at high temperature or at extremes of pH (Kim et al., 1989 ▶). To enhance the stability of monellin, recombinant single-chain monellin proteins were created in which the two natural chains are either directly connected (SCM; Kim et al., 1989 ▶) or joined by a dipeptide linker (MNEI; Tancredi et al., 1992 ▶). These proteins have been characterized by NMR and X-ray crystallography, with five published crystal structures of monellin (Ogata et al., 1987 ▶; Somoza et al., 1993 ▶; Bujacz et al., 1997 ▶; Hung et al., 1998 ▶, 1999 ▶).
Here, we describe a 1.15 Å structure of MNEI. In contrast to all previous X-ray crystal structures of natural or single-chain monellin, MNEI is present in the crystal as a monomer. The substantially higher resolution permits the observation of alternative side-chain conformations and the identification of four negative ions. The latter novel observation is discussed in the context of the proposed electrostatic component of the interaction of sweet proteins with the sweet taste receptor (Esposito et al., 2006 ▶).
2. Experimental
2.1. MNEI amino-acid sequence and nomenclature
MNEI is a single-chain monellin in which chain B and chain A of natural monellin are fused C-terminus to N-terminus, respectively, via a Gly-Phe dipeptide linker. In the original construct (Kim et al., 1989 ▶; Tancredi et al., 1992 ▶), two amino acids (Asn49 and Glu50) were reversed with no effect on the activity (sweetness). To compare our structure with those of natural monellin and SCM, the following conversions are necessary. Amino acids 52–96 of MNEI correspond to chain A (A1–A44) of natural monellin. For SCM, amino acids beyond position 50 correspond to an amino-acid position two higher in MNEI (e.g. Arg70 in SCM is Arg72 in MNEI) owing to the dipeptide linker. We have adopted the nomenclature of Murzin (1993 ▶) for naming the loops between the other secondary-structure elements.
2.2. MNEI expression, purification and crystallization
MNEI was expressed and purified as described previously (Spadaccini et al., 2001 ▶) but using HiPrep16/10 SP FF cation-exchange and HiPrep26/60 Sephacryl 100 columns (GE Healthcare) on an ÄKTA Purifier FPLC. Fractions containing purified protein were pooled, concentrated and dialysed against 10 mM sodium cacodylate pH 5.6. Crystallization conditions were identified using Crystal Screen I (Hampton Research) at 291 K by the hanging-drop vapour-diffusion method with 4 µl drops containing equal volumes of protein (∼10 mg ml−1) and crystallization reagent (0.2 M ammonium sulfate, 30% PEG 4000). After the initial appearance of precipitate, a single crystal of MNEI with approximate dimensions of 0.2 × 0.1 × 0.05 mm grew after 3–4 weeks.
2.3. X-ray data collection, structure determination and refinement
The MNEI crystal was soaked in situ with 40% glycerol solution and flash-frozen in liquid nitrogen. X-ray diffraction data were collected using a Quantum 4 CCD detector (ADSC) at the European Synchrotron Research Facility (ESRF) beamline ID14.1. All data were collected at 100 K, processed using the program MOSFLM (Leslie, 1992 ▶) and sorted and scaled using SORTMTZ, SCALA and TRUNCATE from the CCP4 program package (Collaborative Computational Project, Number 4, 1994 ▶). Although the a and c unit-cell parameters are almost identical, a perfect case for twinning, plots of cumulative intensities output from TRUNCATE show no deviation of observed from theoretical data, indicating that the data is untwinned. The data-collection statistics and experimental parameters are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P21 |
| Resolution (Å) | 33.13–1.15 (1.21–1.15) |
| Unit-cell parameters (Å, °) | a = 27.1, b = 66.3, c = 27.2, α = 90.0, β = 111.6, γ = 90.0 |
| Redundancy | 3.8 (3.7) |
| Total observations | 178910 |
| Unique observations (hkl) | 28254 |
| Completeness (%) | 88.5 (88.5) |
| Rmerge† (%) | 4.3 (25.4) |
| I/σ(I) | 8.9 (2.7) |
| Overall B factor from Wilson plot (Å2) | 9.4 |
| Solvent content (%) | 32.9 |
| Refinement | |
| Resolution limits (Å) | 22.44–1.15 |
| Data cutoff [F/σ(F)] | 0.0 |
| Total No. of reflections | 27957 |
| No. of reflections in working set | 26548 |
| No. of reflections in test set | 1409 |
| Rwork‡ (%) | 13.2 |
| Rfree‡ (%) | 16.2 |
| No. of amino-acid residues | 96 |
| No. of protein atoms | 1074 |
| No. of sulfate ions | 4 |
| No. of water molecules | 143 |
| Average B factor of all atoms (Å2) | 15.6 |
| Average B factor of protein atoms (Å2) | 13.9 |
| Average B factor of solvent atoms (Å2) | 26.6 |
| Ramachandran plot, core (%) | 92.4 |
| Ramachandran plot, allowed (%) | 7.6 |
| R.m.s.d. bond lengths (Å) | 0.026 |
| R.m.s.d. bond angles (°) | 1.99 |
R
merge = 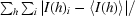
 , where I(h)i is the ith observation of the intensity of reflection h and 〈I(h)〉 is the mean value of all I(h)i.
, where I(h)i is the ith observation of the intensity of reflection h and 〈I(h)〉 is the mean value of all I(h)i.
R = 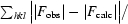
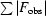 , where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for reflection hkl applied to the work (R
work) and test (R
free) sets, respectively.
, where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for reflection hkl applied to the work (R
work) and test (R
free) sets, respectively.
Initial attempts to solve the structure by the molecular-replacement method with the program MOLREP (Vagin & Teplyakov, 1997 ▶) via the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) were unsuccessful, with several potential solutions producing serious clashes with neighbouring symmetry-related molecules. The data were reindexed into space group P1 to allow the crystal-packing pattern to be observed and to confirm the space group as P21. Specifically, we determined that the MNEI crystal exhibits monomeric packing and not dimeric as observed in all previous monellin crystals. Molecular replacement with MOLREP was then carried out using the 1.8 Å structure of SCM (PDB code 1n98; Hung et al., 1998 ▶) with only one molecule in the asymmetric unit. The molecular-replacement solution was used in rigid-body refinement in the program REFMAC5 (Murshudov et al., 1997 ▶). 5% of the data were randomly selected as a test set to calculate R free and were omitted from all refinements. Two rigid-body domains were defined (residues 10–45 in chain B and 55–90 in chain A) and the full resolution range of reflections from 33.13 to 1.15 Å was used. Starting R factors were 46.0% for R work and 49.5% for R free. σA-weighted 2|F o − F c| and |F o − F c| difference maps were generated and inspected by eye in the program O (Jones et al., 1991 ▶) and were found to be of good quality. The model was then refined with CNS (Brünger et al., 1998 ▶) by simulated annealing to reduce model bias, starting with an initial temperature of 3000 K. At this stage, the model was manually corrected by inspection of maps. The model was then refined with REFMAC5 using Babinet scaling and after several iterations of maximum-likelihood restrained refinement and rebuilding, water molecules were introduced gradually using the ARP_waters command in conjunction with map inspection. Any molecules corresponding to 2|F o − F c| peaks of less than 3σ or lying further than 3.5 Å from all potential hydrogen-bonding partners were deleted. H atoms were added in the riding positions, temperature factors were refined anisotropically and the model was refined to convergence. The final model from this stage of refinement contained 911 non-H atoms, corresponding to the 96 amino acids of MNEI and 114 water molecules, with an R work and R free of 16.7% and 20.2%, respectively. Further visual inspection of maps using the program Coot (Emsley & Cowtan, 2004 ▶) indicated numerous patches of positive density near various main-chain atoms and well defined side-chain atoms that were not obvious in previous maps. These were particularly prominent near the side-chain atoms of Asp7, Asp21, Lys36, Cys41, Met42 and Arg72 and were subsequently modelled as alternative side chains. A further 32 alternative conformations were identified in subsequent rounds of refinement and model building using REFMAC5 and Coot, respectively. All were modelled with half occupancy unless the B factors indicated otherwise and were refined using anisotropic B factors for all atoms including solvent. The alternative conformations for each residue are denoted A and B following the residue number. Rebuilding was aided by systematically checking all electron-density peaks greater than 5σ in the |F o − F c| Fourier maps using the Difference Map Peaks command in Coot. A total of 143 waters and four sulfate ions were identified and fitted accordingly, giving final R work and R free values of 13.2% and 16.2%, respectively. Refinement statistics are also shown in Table 1 ▶. Ramachandran plots (Ramachandran & Sasisekharan, 1968 ▶) were assessed using PROCHECK (Laskowski et al., 1993 ▶) and indicate that the final model is of high quality (Table 1 ▶).
3. Results and discussion
3.1. Quality of the protein structure
Monellin has a secondary structure consisting of five β-strands that form an antiparallel β-sheet (β1–β5) and a 17-residue α-helix (α1) cradled in the concave face of strands β2–β5 (Fig. 1 ▶). In this structure, like some others (Lee et al., 1999 ▶; Sung et al., 2001 ▶), a short β-strand flanked by two loop regions is identified between the α-helix and β2. For consistency, we have named this β2a and retained the numbering of the other strands used elsewhere. In MNEI and SCM the β-sheets are joined by three main loops: β2 and β3 are joined by the engineered loop L23 (residues 47–56), β3 and β4 are joined by a short left-handed loop L34 (residues 66 to 69) and β4 and β5 are connected by a short right-handed loop L45 (residues 78–82). The polypeptide chain ends with a short sequence containing four proline residues; three of these, Pro94–Pro96, form a 310-polyproline II helix, the presence of which was confirmed by circular-dichroism spectroscopy (results not shown). The MNEI structure contains two cis-prolines: Pro40, located at the start of β3 on the ‘convex’ side of the protein, and Pro92 at the C-terminus.
Figure 1.

MNEI structure and crystal packing. (a) Cartoon of the MNEI structure in two approximately orthogonal views. The β-sheets and loops are labelled as described in the main text (L23 contains the Gly-Phe dipeptide linker used to fuse the two chains of natural monellin). (b) Molecular packing in the MNEI crystal with a single asymmetric unit shown.
The electron density for the polypeptide chain is unambiguous for almost all residues, resulting in a high-quality final model. The only exception is for residues 78–81 (main-chain atoms in loop L45), where there is some disorder visible in the |F o − F c| maps. Of the 96 residues, 92.6% are in the core region of the Ramachandran plots and 7.4% are in the allowed region. The 38 alternate conformations were modelled unambiguously, with the exception of the C-terminal proline residues 94–96. Although alternate conformations were modelled for this region, there is still some indication of disorder. A 4.4σ negative |F o − F c| peak (corresponding to 0.29 e Å−3) was observed at the position of the hydroxyl group of Tyr79. Removal of the hydroxyl group eliminates this peak and does not result in significant positive |F o − F c| density. This is most likely to indicate specific radiation damage (Burmeister, 2000 ▶) or possibly electron redistribution in this area of the structure owing to the high mobility of the loop.
3.2. Crystal packing and surface contacts
Despite exhibiting many crystal forms (Ogata & Kim, 1986 ▶; Ogata et al., 1987 ▶; Somoza et al., 1993 ▶; Bujacz et al., 1997 ▶; Hung et al., 1997 ▶, 1998 ▶, 1999 ▶), previous natural monellin and SCM crystals have always contained two proteins positioned in such a way as to suggest that a dimeric complex may be present (comprising two copies of both chain A and chain B in the case of natural monellin). The MNEI crystal we describe here belongs to a space group that has previously been observed for monellin (P21; Somoza et al., 1993 ▶), but with markedly different unit-cell parameters and resolution limits than those of SCM (PDB code 1mol; unit-cell parameters a = 46.4, b = 49.0, c = 40.7 Å, α = γ = 90, β = 102.9°, resolution limit 1.7 Å) and natural monellin (3mon; unit-cell parameters a = 39.8, b = 87.2, c = 72.1 Å, α = β = 90, γ = 107.3°, resolution limit 2.75 Å). Most strikingly, our MNEI structure displays a completely new crystal-packing arrangement: a single monellin molecule is contained in the asymmetric unit (compared with two and four molecules in the asymmetric unit for 1mol and 3mon, respectively) and no dimer interface is observed (Fig. 1 ▶). This monomeric crystalline form of MNEI provides new validation of the monellin structure and supports evidence from solution studies (Lee et al., 1999 ▶; Spadaccini et al., 2001 ▶) that the functional unit of the protein is the monomer (i.e. single heterodimer of chain A and chain B in natural monellin).
The close-packed nature of the MNEI crystal results in a greater number of contacts between symmetry-related molecules, including several made by residues of the ‘flexible’ loop L23 (Tyr47–Glu54). Interestingly, this is the only site of sequence variation between MNEI and SCM. The addition of the GF dipeptide linker to this loop and the altered contacts it makes as a result appear to play a significant role in producing this new crystal form.
The MNEI crystal contains small (∼11 Å wide) but clearly defined solvent channels (Fig. 1 ▶) and displays remarkably compact packing as indicated by the Matthews coefficient (V M; Matthews, 1968 ▶) of 1.8 Å3 Da−1. In contrast, the 1mol and 3mon structures are more loosely packed, with V M values of 2.1 and 2.4 Å3 Da−1, respectively. This compact packing arrangement and the associated low solvent content (33%) of this MNEI crystal is most likely to be the reason for its atomic resolution. It is also probable that this low solvent content has played a part in the identification of alternate conformations, as packing forces can stabilize alternate conformations in protein crystals (Zhang et al., 1995 ▶).
This structure reveals the lowest B factors of all published crystal structures of monellin and this, together with the lower water content and increased crystal contacts, can be interpreted as a lower flexibility of MNEI in this monomeric form. The loop sections in this structure and in 1mol and 3mon do, however, show a high degree of static disorder, i.e. higher mean B factors. To compare our structure with previously determined crystal structures of 3mon, orthorhombic natural monellin (PDB code 4mon; Bujacz et al., 1997 ▶) and 1mol, pairwise superpositions were made using the Cα atoms of residues 1–46 and 57–96 (the intrinsically flexible residues located on L23 were excluded). The low root-mean-square deviation (r.m.s.d.) values obtained (Table 2 ▶) indicate that, as expected, each of these structures is globally very similar.
Table 2. Root-mean-square deviation (r.m.s.d.) of backbone atoms for residues 10–40 and 58–94 between MNEI and previous crystal and NMR structures of natural and single-chain monellin proteins.
3.3. Solvent structure
The final refined model includes 143 water molecules (131 full occupancy, three at half occupancy and nine dual occupancy). The isotropic B factors for these water molecules are in the range 9–57 Å2, with an average of 26 Å2. This MNEI structure and the previous highest resolution structure of 1mol were aligned using the Secondary Structure Matching (SSM) command (Krissinel & Henrick, 2004 ▶) in Coot with all water molecules included. The aligned structures were analysed using the CCP4 program CONTACT to identify conserved water sites, with the contact-distance criterion set at 0.0–1.0 Å (Zhang & Matthews, 1994 ▶). B factors for the identified waters were also assessed and a value of <40 Å2 was taken to be consistent with them being stably bound. This analysis identified 20 conserved water molecules between the two models (Table 3 ▶). Of these, two sites are completely buried within the protein. The first (W1) is located between sheets β2 and β3, immediately behind the α-helix, and forms hydrogen bonds with the main-chain atoms of Ile38 (O), Pro40 (O) and Tyr63 (N). An additional interaction is seen with the S atom of Cys41B in this MNEI structure only. The second water site (W17) is located at the base of the α-helix on Lα2 and forms hydrogen bonds with main-chain atoms of Glu23 (O), Tyr29 (N) and Gly30 (O). Additional interactions with all the main-chain atoms of Gly27 and Gln28 (N) are only seen in our MNEI structure.
Table 3. Conserved and buried waters within 10 Å of each other.
| MNEI | SCM (1mol) | Location |
|---|---|---|
| 1 | 309 | Buried |
| 2 | 311 | Surface |
| 4 | 410 | Surface |
| 7 | 384 | Surface |
| 12 | 431 | Surface |
| 17 | 302 | Buried |
| 22 | 346 | Surface |
| 23 | 320 | Surface |
| 32 | 362 | Surface |
| 33 | 324 | Surface |
| 50 | 463 | Surface |
| 61 | 573 | Surface |
| 91 | 421 | Surface |
| 97 | 349 | Surface |
| 103 | 359 | Surface |
| 104 | 348 | Surface |
| 120 | 478 | Surface |
| 122 | 307 | Surface |
| 127 | 328 | Surface |
| 128 | 575 | Surface |
Four stably bound ions were also located, each with the characteristic tetrahedral electron density of a sulfate or phosphate ion. Both anions were present during protein purification (phosphate) or crystallization (sulfate), but given the vastly greater sulfate concentration in the crystallization conditions, each site was modelled as a sulfate ion (S1–S4). These observations represent the first instance of negative ions bound to a sweet protein and may provide insight into potential electrostatic protein–protein interactions. Three of these ions are located on the concave face of the protein, with the fourth (S3) on the opposite face (Fig. 2 ▶). The first site (S1) interacts with Glu48, Arg53, Lys56, Asp78 and three water molecules. S2 is bound by residues Arg39 and Asn49, four water molecules and by Arg31 from a symmetry-related molecule. S4 interacts with Arg53, Phe52 and three waters and Asn14B, Lys17 (A and B) and a further two waters from a symmetry-related molecule. The final sulfate ion (S3), located on the convex side of the protein, is bound by Arg72, Pro94 (A and B) and one water molecule and has symmetry-related contacts to Glu22 (A and B) and a further three water molecules. This MNEI–ion interaction is of particular interest as the ion lies adjacent to a patch of positive surface potential and is surrounded by residues identified as being important for MNEI sweetness (Fig. 2 ▶). The importance of charge complementarity between the largely positive monellin surface and negative T1R2–T1R3 complex for sweet protein–receptor interaction was highlighted by a recent study (Esposito et al., 2006 ▶) and the present structure suggests that the surface of MNEI in the vicinity of S3 is a potential site of such an electrostatic interaction.
Figure 2.

Surface representation of MNEI and bound ions. (a) Electrostatic potential of the concave (left) and convex (right) MNEI surfaces with bound sulfate ions (S1–S4). Positive and negative surface potential is shown in blue and red, respectively. (b) Surface representation of MNEI, highlighting key residues for sweetness (shown in the same orientation as in a). Residues important for putative charge–charge interactions between MNEI and the sweet taste receptor T1R2–T1R3 are shown in green and other key residues for MNEI sweetness are shown in blue and violet (where mutation causes a decrease in sweetness of one and two orders of magnitude, respectively).
3.4. Discrete disorder
The quality of the 1.15 Å electron-density map allowed the side chains of 38 residues (Table 4 ▶) to be modelled in two conformations. Although most of these occur on the surface of the protein, Thr12, Cys41, Leu62 and Ile75 are found in the hydrophobic core. The majority of the disordered residues were found to interact through hydrogen-bonding networks, either directly or indirectly via water molecules, to other disordered regions in the protein. The most striking example is located in a patch on the convex side of the protein that has been suggested to participate in binding to the T1R2–T1R3 sweet taste receptor (Morini & Temussi, 2005 ▶; Temussi, 2006 ▶). Here, a network of 15 discrete side-chain conformations can be traced (Fig. 3 ▶), consisting of the A or B conformations of Glu4, Ile5, Ile6, Asp7, Cys41, Met42, Glu59, Gln61, Tyr63, Arg72, Asp74, Arg88 and the C-terminal prolines 94–96. The most obvious changes to side-chain conformation occur in residues Arg72 and Arg88, where the entire side chain has rotated away from the body of the protein and out towards the solvent space. Several of these residues observed in double conformations in this structure are thought to be important in conferring sweet taste to the protein (Kohmura et al., 1992a ▶,b ▶; Ariyoshi, 1994 ▶; Somoza et al., 1995 ▶; Niccolai et al., 2001 ▶). These discretely disordered residues may provide the structural plasticity that enables monellin to interact and optimize its large surface complementarity with the sweet taste receptor according to the ‘wedge’ model (Tancredi et al., 2004 ▶).
Table 4. MNEI residues modelled with alternate conformations.
Residues highlighted in bold have been shown to be important for conferring sweet taste to monellin.
| Residues | Glu4, Ile5, Ile6, Asp7, Phe11, Thr12, Gln13, Asn14, Lys17, Asp21, Glu22, Glu23, Asn24, Lys25, Gln28, Lys36, Cys41, Met42, Ile46, Glu54, Ile55, Lys56, Glu59, Gln61, Leu62, Tyr63, Lys69, Arg72, Asp74, Ile75, Glu77, Arg84, Lys85, Arg88, Pro92, Pro94, Pro95, Pro96 |
Figure 3.

Stereo representation of discrete disorder in the MNEI structure. An extended region of disordered residues on the convex (putative receptor-binding) side of protein contains a network of 15 discrete side-chain conformations (shown in blue and red).
4. Conclusion
The X-ray crystal structure of MNEI, a single-chain version of the sweet protein monellin, was determined and refined at 1.15 Å resolution. Despite exhibiting a distinctly different packing arrangement and crystal contacts, the global fold of the protein is essentially identical to previous structures, providing further validation of the known structure of monellin. This crystal is unique as it lacks the dimer interface seen in all previous crystal structures of monellin and its single-chain derivatives. This new crystal form shows that monellin can exist as a monomer in both the crystalline and solution states (Morris et al., 1973 ▶; Lee et al., 1999 ▶) and further confirms that this is the functional unit of the protein for interaction with the T1R2–T1R3 sweet taste receptor.
Our high-resolution model, with numerous side chains in dual conformations and bound negative ions, provides useful information for further understanding the nature of the MNEI–T1R2–T1R3 interaction. Many of the key residues for monellin sweetness and those implicated in electrostatic interaction are located in a patch on the convex side of the protein near a bound sulfate ion and several are observed in two discrete conformations. To distinguish the role(s) of these key residues with regard to their involvement in binding to the sweet taste receptor will require further detailed structural analysis of monellin interactions with the sweet taste receptor.
Supplementary Material
PDB reference: monellin, 2o9u, r2o9usf
Acknowledgments
We thank P. A. Temussi for providing the MNEI expression vector, S. L. Allinson and C. M. Dunham for assistance with X-ray data collection and the staff of the European Synchrotron Radiation Facility (ESRF) beamline ID14.1. Crystallographic figures were generated using PyMOL (DeLano & Lam, 2005 ▶). This work was supported by funding from the National Institute on Deafness and Other Communication Disorders, National Institutes of Health (DC05786).
References
- Ariyoshi, Y. (1994). J. Synth. Org. Chem. Jpn, 52, 359–369.
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bujacz, G., Miller, M., Harrison, R., Thanki, N., Gilliland, G. L., Ogata, C. M., Kim, S.-H. & Wlodawer, A. (1997). Acta Cryst. D53, 713–719. [DOI] [PubMed] [Google Scholar]
- Burmeister, W. P. (2000). Acta Cryst. D56, 328–341. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- DeLano, W. L. & Lam, J. W. (2005). Abstr. Pap. Am. Chem. Soc.230, U1371–U1372.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Esposito, V., Gallucci, R., Picone, D., Saviano, G., Tancredi, T. & Temussi, P. A. (2006). J. Mol. Biol.360, 448–456. [DOI] [PubMed] [Google Scholar]
- Hellekant, G., Glaser, D., Brouwer, J. N. & Vanderwel, H. (1976). Acta Physiol. Scand.97, 241–250. [DOI] [PubMed] [Google Scholar]
- Hellekant, G., Hladik, C. M., Dennys, V., Simmen, B., Roberts, T. W., Glaser, D., Dubois, G. & Walters, D. E. (1993). Chem. Senses, 18, 307–320.
- Hung, L. W., Kohmura, M., Ariyoshi, Y. & Kim, S.-H. (1997). Acta Cryst. D53, 327–328. [DOI] [PubMed] [Google Scholar]
- Hung, L. W., Kohmura, M., Ariyoshi, Y. & Kim, S.-H. (1998). Acta Cryst. D54, 494–500. [DOI] [PubMed] [Google Scholar]
- Hung, L. W., Kohmura, M., Ariyoshi, Y. & Kim, S.-H. (1999). J. Mol. Biol.285, 311–321. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kim, S.-H., Kang, C. H., Kim, R., Cho, J. M., Lee, Y. B. & Leg, T. K. (1989). Protein Eng.2, 571–575. [DOI] [PubMed] [Google Scholar]
- Kohmura, M., Nio, N. & Ariyoshi, Y. (1992a). Biosci. Biotechnol. Biochem.56, 472–476. [DOI] [PubMed] [Google Scholar]
- Kohmura, M., Nio, N. & Ariyoshi, Y. (1992b). Biosci. Biotechnol. Biochem.56, 1937–1942. [DOI] [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Lee, S. Y., Lee, J. H., Chang, H. J., Cho, J. M., Jung, J. W. & Lee, W. T. (1999). Biochemistry, 38, 2340–2346. [DOI] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Morini, G. & Temussi, P. A. (2005). Chem. Senses, 30, i86–i87. [DOI] [PubMed] [Google Scholar]
- Morris, J. A. & Cagan, R. H. (1972). Biochim. Biophys. Acta, 261, 114–122. [DOI] [PubMed] [Google Scholar]
- Morris, J. A., Martenson, R., Deibler, G. & Cagan, R. H. (1973). J. Biol. Chem.248, 534–539. [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Murzin, A. G. (1993). J. Mol. Biol.230, 689–694. [DOI] [PubMed] [Google Scholar]
- Niccolai, N., Spadaccini, R., Scarselli, M., Bernini, A., Crescenzi, O., Spiga, O., Ciutti, A., Di Maro, D., Bracci, L., Dalvit, C. & Temussi, P. A. (2001). Protein Sci.10, 1498–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, C., Hatada, M., Tomlinson, G., Shin, W. C. & Kim, S.-H. (1987). Nature (London), 328, 739–742. [DOI] [PubMed] [Google Scholar]
- Ogata, C. M. & Kim, S.-H. (1986). Biophys. J.49, A438–A438.
- Ramachandran, G. N. & Sasisekharan, V. (1968). Adv. Protein Chem.23, 283–437. [DOI] [PubMed] [Google Scholar]
- Somoza, J. R., Cho, J. M. & Kim, S.-H. (1995). Chem. Senses, 20, 61–68. [DOI] [PubMed] [Google Scholar]
- Somoza, J. R., Jiang, F., Tong, L., Kang, C. H., Cho, J. M. & Kim, S.-H. (1993). J. Mol. Biol.234, 390–404. [DOI] [PubMed] [Google Scholar]
- Spadaccini, R., Crescenzi, O., Tancredi, T., De Casamassimi, N., Saviano, G., Scognamiglio, R., Di Donato, A. & Temussi, P. A. (2001). J. Mol. Biol.305, 505–514. [DOI] [PubMed] [Google Scholar]
- Sung, Y. H., Hong, H. D., Cheong, C., Kim, J. H., Cho, J. M., Kim, Y. R. & Lee, W. (2001). J. Biol. Chem.276, 44229–44238. [DOI] [PubMed] [Google Scholar]
- Tancredi, T., Iijima, H., Saviano, G., Amodeo, P. & Temussi, P. A. (1992). FEBS Lett.310, 27–30. [DOI] [PubMed] [Google Scholar]
- Tancredi, T., Pastore, A., Salvadori, S., Esposito, V. & Temussi, P. A. (2004). Eur. J. Biochem.271, 2231–2240. [DOI] [PubMed] [Google Scholar]
- Temussi, P. A. (2006). Cell. Mol. Life Sci.63, 1876–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Zhang, X. J. & Matthews, B. W. (1994). Protein Sci.3, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. J., Wozniak, J. A. & Matthews, B. W. (1995). J. Mol. Biol.250, 527–552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: monellin, 2o9u, r2o9usf