

Abstract
The Hsp70 molecular chaperones are ATPases that play critical roles in the pathogenesis of many human diseases, including breast cancer. Hsp70 ATP hydrolysis is relatively weak, but is stimulated by J domain-containing proteins. We identified pyrimidinone-peptoid hybrid molecules that inhibit cell proliferation with greater potency than previously described Hsp70 modulators. In many cases, anti-proliferative activity correlated with inhibition of J domain stimulation of Hsp70.
1. Introduction
The heat shock protein Hsp70, and its constitutively expressed counterpart Hsc70, play essential roles in many diverse cellular processes, including protein folding and transport, and the rearrangement of multi-protein complexes1–3. Hsp70 contains an N-terminal ATPase domain and a C-terminal peptide binding domain4–6. Hsp70 substrates bind the peptide binding domain and stimulate the ATPase activity of Hsp70. However, Hsp70 is a weak ATPase and maximal Hsp70 activity requires J domain-containing proteins, such as the Hsp40 class of molecular chaperones7. The J domain interacts with the Hsp70 ATPase domain through a conserved His-Pro-Asp sequence to stimulate Hsp70 ATP hydrolysis8,9. One example of a J domain-containing protein is the large tumor antigen (TAg) of the polyomavirus, Simian Virus 40 (SV40). SV40 was a contaminant of the original polio vaccine and transforms rodent cell lines in culture. The TAg J domain can stimulate Hsp70 ATP hydrolysis, an activity that is required for maximal tumorigenesis10. SV40 is highly related to at least four human polyomaviruses11, 12, two of which have been definitively linked to two disease states—progressive multifocal leukoencephalopathy (PML) and kidney transplant rejection—in immunocompromised patients13.
Hsp70 has been reported to be upregulated in various cancers including breast, lung, colon, and cervix14–16. However, it is unclear if changes in expression lead to tumor formation or if these changes are in response to the tumor environment. In support of the first scenario, the over-expression of Hsp70 is sufficient to transform some cell lines, and transgenic mice constitutively over-expressing Hsp70 develop T-cell lymphomas17, 18. Consistent with these data, reduction of Hsp70 levels by antisense RNA is sufficient to induce apoptosis in several breast cancer cell lines, and Hsp70 over-expression in breast cancer tissues has been correlated with increased resistance to chemotherapy and poor prognosis19–22.
Until recently, 15-deoxysperagulin (DSG) was the only Hsp70 modulator reported23. DSG has been used clinically as an immunosuppressant to prevent kidney rejection after transplantation, and ~70% of kidney transplant patients who were treated with DSG showed an excellent or good response24. However, current studies show that other immunosuppressive regimens have a lower rate of kidney rejection25, and therefore DSG is not widely used clinically. In in vitro experiments DSG interacts with similar affinities (KD = ~4–5µM) with both Hsp70 and Hsp9026. Moreover, the mechanism by which DSG affects chaperone function is unclear, although the compound modestly stimulates Hsp70’s ATPase activity27 and may bind to the C-terminus of Hsp7028, a region involved in co-chaperone interaction. Based on the above noted limitations of DSG, there is a need for more specific Hsp70 modulators with defined modes of action that can be used as biological probes and also, potentially, as improved therapeutic agents.
To this end, we sought and previously reported on structural analogs of DSG that affect Hsp70 ATPase activity29, 30. These efforts led to the identification of compounds such as MAL3-101 (Figure 1) that interfere with TAg stimulation of Hsp70’s ATPase activity29. Compounds that interfere with J domain stimulation of Hsp70 may also be anti-tumorigenic since Hsp70 must interact with specific J domain-containing proteins to perform each of its many functions31, 32. In fact, MAL3-101 induces apoptosis in the SK-BR-3 breast cancer cell line with an apparent ED50 of <10 µM33. Unfortunately, MAL3-101 has a high molecular weight, low solubility, and high lipophilicity. In contrast, chemical derivatives of MAL3-101 might exhibit improved physical characteristics while still maintaining or possessing improved anti-proliferative and chaperone modulating activities.
Figure 1.
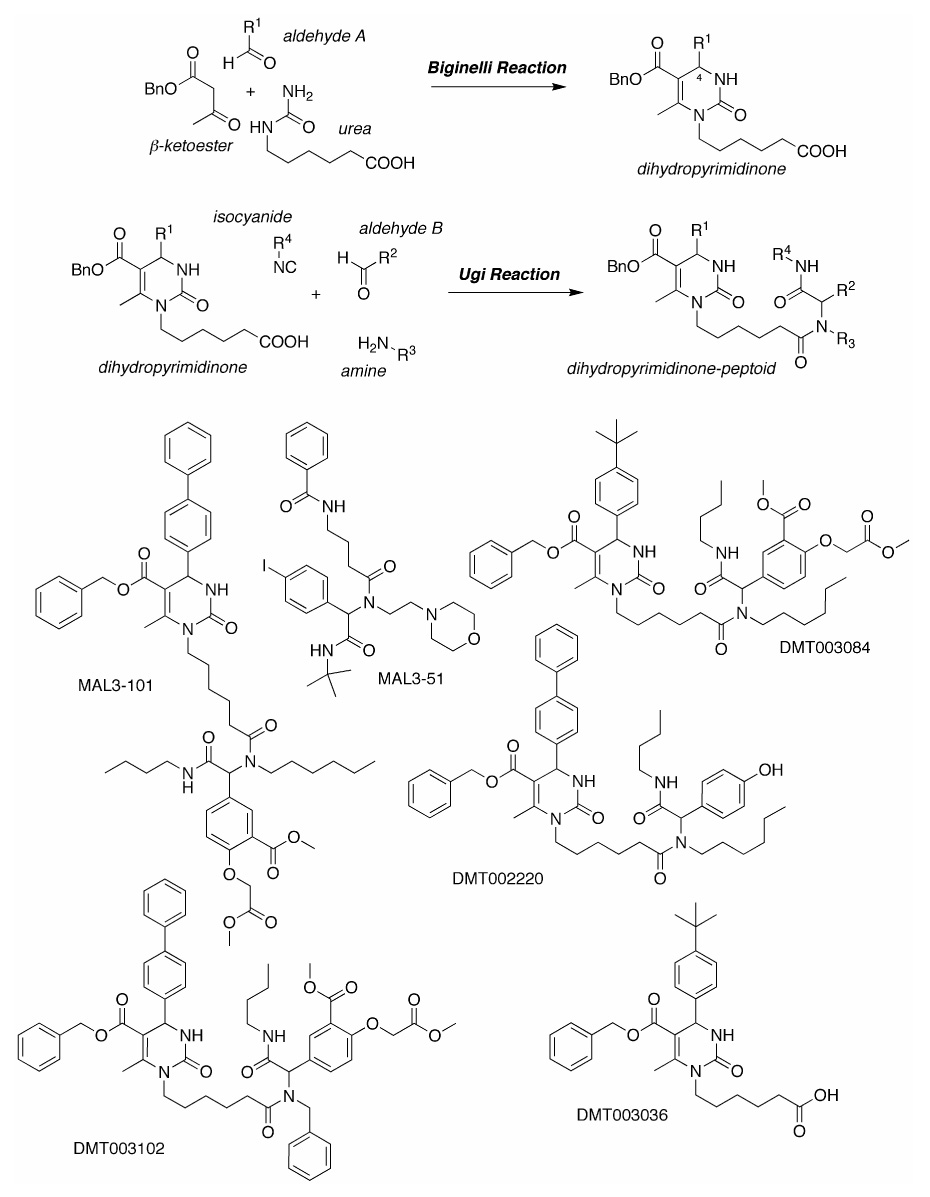
Synthetic sequence and representative products
We now report on the synthesis and characterization of new MAL3-101 derivatives. These compounds, and previously described compounds with similar characteristics34, were screened for their effects on SK-BR-3 breast cancer cell proliferation, on endogenous Hsp70 ATPase activity, and on TAg (J domain) - stimulated ATPase activity. We discovered that members of two derivative classes inhibited breast cancer cell growth in the low micromolar range, some at lower concentrations than MAL3-101. In many cases, these effects correlated with inhibition of TAg-stimulated Hsp70 ATP hydrolysis. Our study is the first to relate distinct small molecule compounds to Hsp70 modulation and reduced breast cancer cell proliferation.
2. Results and Discussion
Since MAL3-101 induces apoptosis in breast cancer cells33, a library of structurally related compounds was prepared in an effort to identify those with improved physiochemical, biochemical and cellular activities. A total of 27 analogs of MAL3-10134 were synthesized, analogous to MAL3-101, through successive three-component Biginelli and four-component Ugi condensation reactions to create pyrimidinone-peptoid hybrid (referred to as dihydropyrimidinones in reference 29) (Figure 1). These compounds were placed in three distinct classes depending on the specific reagents utilized in the reactions (see Table 1). Substituents on the pyrimidinone at R1 are in Class 3, whereas modifications of R2 and R3 of the peptoid are in Classes 4 and 5, respectively. An additional 15 compounds, precursors to the MAL3-101 analogs, were isolated by the Biginelli reaction without a subsequent Ugi reaction34 and were placed in Class 1 and 2 (Table 1; Class 2 compounds are methyl ester derivatives of distinct compounds in Class 1). The free diacid of MAL3-101 (Class 6) and a derivative of a related compound, MAL3-39 (Class 7), were also synthesized. The specific chemical modifications of each compound are shown in Table 1 and the structures of select compounds are shown in Figure 1.
Table 1.
Substituents utilized to create pyrimidinone-peptoid hybrid molecule
| Class | Name | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|
| N/A | MAL3-101 | p-biphenyl | 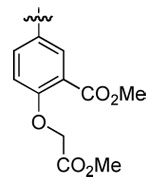 |
n-hexyl | n-butyl |
| N/A | MAL3-51 | Different scaffold | p-iodophenyl | 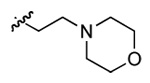 |
t-butyl |
| 1 | DMT002272 | o-biphenyl | Different scaffold | ||
| 1 | DMT003034 | c-hexyl | Different scaffold | ||
| 1 | DMT003036 | p-(t-buyl)phenyl | Different scaffold | ||
| 1 | DMT003038 | c-propyl | Different scaffold | ||
| 1 | DMT003116 | p-(c-hexyl)phenyl | Different scaffold | ||
| 1 | MAL1-274 | p-chlorophenyl | Different scaffold | ||
| 1 | MAL2-06A | 2-naphthyl | Different scaffold | ||
| 1 | MAL2-10A | phenyl | Different scaffold | ||
| 1 | MAL2-116-17 | m-nitrophenyl | Different scaffold | ||
| 1 | MAL2-116-20 | p-trifluoromethylphenyl | Different scaffold | ||
| 1 | MAL2-11B | p-biphenyl | Different scaffold | ||
| 1 | MAL2-13 | p-nitrophenyl | Different scaffold | ||
| 2 | DMT003042* | p-biphenyl | Different scaffold | ||
| 2 | DMT003044* | phenyl | Different scaffold | ||
| 2 | DMT003047* | c-propyl | Different scaffold | ||
| 3 | DMT003082 | p-trifluoromethylphenyl | 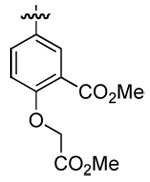 |
n-hexyl | n-butyl |
| 3 | DMT003084 | p-(t-buyl)phenyl | 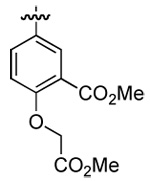 |
n-hexyl | n-butyl |
| 3 | DMT003086 | m-nitrophenyl | 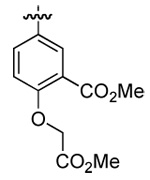 |
n-hexyl | n-butyl |
| 3 | DMT003088 | phenyl | 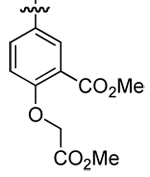 |
n-hexyl | n-butyl |
| 3 | DMT003090 | p-chlorophenyl | 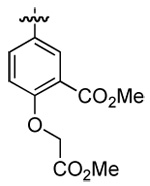 |
n-hexyl | n-butyl |
| 3 | DMT003092 | p-nitrophenyl | 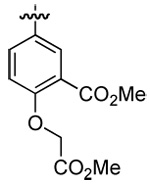 |
n-hexyl | n-butyl |
| 3 | DMT003094 | 2-naphthyl | 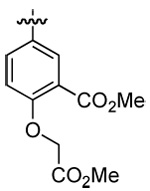 |
n-hexyl | n-butyl |
| 3 | DMT003096 | o-biphenyl | 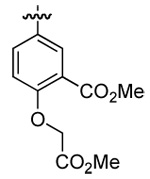 |
n-hexyl | n-butyl |
| 3 | DMT003100 | c-hexyl | 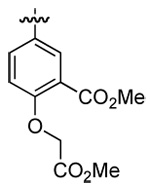 |
n-hexyl | n-butyl |
| 3 | DMT003132 | c-propyl | 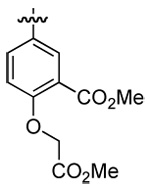 |
n-hexyl | n-butyl |
| 3 | DMT003134 | p-(c-hexyl)phenyl | 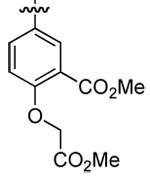 |
n-hexyl | n-butyl |
| 4 | DMT002218 | p-biphenyl | phenyl | n-hexyl | n-butyl |
| 4 | DMT002220 | p-biphenyl | p-hydroxyphenyl | n-hexyl | n-butyl |
| 4 | DMT002222 | p-biphenyl | p-biphenyl | n-hexyl | n-butyl |
| 4 | DMT002260 | p-biphenyl | 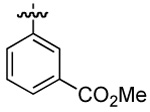 |
n-hexyl | n-butyl |
| 4 | DMT002262 | p-biphenyl | methyl | n-hexyl | n-butyl |
| 4 | DMT002264 | p-biphenyl | c-hexyl | n-hexyl | n-butyl |
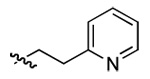
| 4 | DMT003058 | p-biphenyl | 2-pyridyl | n-hexyl | n-butyl |
| 4 | DMT003112 | p-biphenyl | 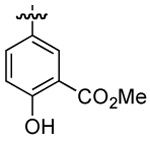 |
n-hexyl | n-butyl |
| 4 | DMT003114 | p-biphenyl | 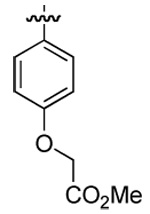 |
n-hexyl | n-butyl |
| 5 | DMT002286 | p-biphenyl | 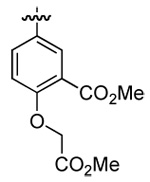 |
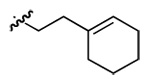 |
n-butyl |
| 5 | DMT003052 | p-biphenyl | 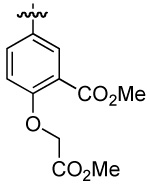 |
n-butyl | |
| 5 | DMT003102 | p-biphenyl | 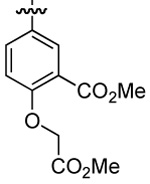 |
benzyl | n-butyl |
| 5 | DMT003104 | p-biphenyl | 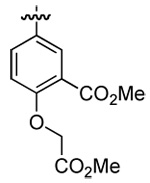 |
2-phenethyl | n-butyl |
| 5 | DMT003106 | p-biphenyl | 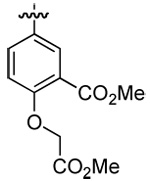 |
 |
n-butyl |
| 5 | DMT003108 | p-biphenyl | 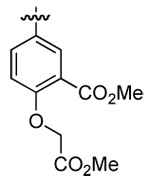 |
 |
n-butyl |
| 5 | DMT003110 | p-biphenyl | 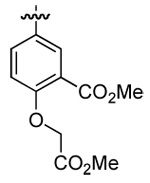 |
 |
n-butyl |
| 6 | DMT003020 | p-biphenyl | 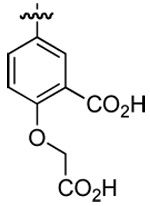 |
n-hexyl | n-butyl |
| 7 | DMT003024 | p-nitrophenyl | p-biphenyl | 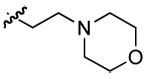 |
n-butyl |
| N/A | PTX(nM) | Different scaffold |
denotes a methyl ester
Initially, all compounds were screened for inhibition of SK-BR-3 breast cancer cell growth in an established cell proliferation assay35, 36. The concentration of compound at which cellular growth is inhibited to 50% of the DMSO control (GI50) was calculated. In this assay, the GI50 for MAL3-101 was 27 ± 2 µM (Table 2), which agrees well with the effect of this compound on induction of the apoptotic pathway in SK-BR-3 cells33. MAL3-51 (Figure 1), a structurally related compound with little effect on chaperone function29, had a significantly less pronounced activity (Table 2). Paclitaxel, a small molecule stabilizer of microtubules that stimulates cellular apoptosis37, 38, was used as a positive control and inhibited breast cancer cell proliferation with a GI50 of 6.1 ± 1.2 nM (“PTX”, Table 2). Notably, a number of MAL3-101 derivatives inhibited SK-BR-3 cell growth; GI50s for sixteen compounds ranged from 6.0 ± 0.4 µM to 42.6 ± 6 µM, with nine compounds yielding GI50 values < 10 µM. We then examined the effects of select compounds with activity in this assay on the growth of two other cell lines: MCF7 breast cancer cells and HT29 colon cancer cells. DMT003088 and DMT003132 (Class 3) and DMT003052 (Class 5) exhibited GI50s in SK-BR-3 cells at a range from 6.2–8.8 µM, and similarly inhibited MCF7 and HT29 growth with GI50s ranging from 2.4–7.8 µM (Table 3). Of interest, MAL3-101 also inhibited MCF7 growth but had no effect on HT29 cells. This result suggests that MAL3-101 exhibits some specificity of action with regard to different cancer cell lines, as previously proposed33.
Table 2.
In vivo and in vitro effects of the pyrimidinone-peptoid compounds. The GI50 for each compound (in µM) was assayed as described in the Experimental Methods as was the fold change for the endogenous and TAg-stimulated Hsp70 ATPase activities.
| Name | SK-BR-3 GI50 | Fold Change Hsp70 ATP Hydrolysis | Fold Change TAg-Stimulated Hsp70 ATP Hydrolysis |
|---|---|---|---|
| MAL3-101 | 27±2 | 1.7 | −2.0 |
| MAL3-51 | >50 | 1.1 | −2.0 |
| DMT002272 | >50 | 1.0 | −5.0 |
| DMT003034 | >50 | 1.0 | 1.5 |
| DMT003036 | >50 | 1.0 | −3.5 |
| DMT003038 | >50 | 1.1 | 1.0 |
| DMT003116 | >50 | 1.1 | −2.4 |
| MAL1-274 | >50 | 1.3 | −1.6 |
| MAL2-06A | >50 | −1.1 | −1.4 |
| MAL2-10A | >50 | 1.5 | −1.1 |
| MAL2-116-17 | >50 | −1.1 | 1.1 |
| MAL2-116-20 | >50 | 1.2 | 1.1 |
| MAL2-11B | >50 | 1.1 | −3.5 |
| MAL2-13 | >50 | 1.2 | 1.1 |
| DMT003042 | >50 | 2.6 | −1.2 |
| DMT003044 | >50 | 2.3 | −1.7 |
| DMT003046 | >50 | 1.9 | 1.0 |
| DMT003082 | >50 | 1.0 | −4.8 |
| DMT003084 | 29±0 | 1.2 | −5.2 |
| DMT003086 | 6.0±0.4 | 1.1 | −1.5 |
| DMT003088 | 6.5±0.7 | 1.1 | −1.1 |
| DMT003090 | 16±6 | 1.3 | −5.1 |
| DMT003092 | 6.9±0.4 | 1.0 | −1.1 |
| DMT003094 | 10±1 | 1.0 | −1.1 |
| DMT003096 | 17±3 | −1.1 | −4.0 |
| DMT003100 | 6.3±1.0 | 1.2 | −2.1 |
| DMT003132 | 6.2±0.4 | 1.2 | −2.6 |
| DMT003134 | 42±6 | 2.2 | −1.6 |
| DMT002218 | >50 | 3.2 | 1.1 |
| DMT002220 | >50 | 11.8 | 9.2 |
| DMT002222 | >50 | 9.9 | 6.0 |
| DMT002260 | >50 | 3.7 | −2.0 |
| DMT002262 | >50 | 1.6 | 1.0 |
| DMT002264 | >50 | 3.2 | 1.1 |
| DMT003058 | >50 | 4.2 | 5.5 |
| DMT003112 | >50 | 19.1 | 2.1 |
| DMT003114 | 48±5 | 1.1 | 1.2 |
| DMT002286 | 39±6 | 6.1 | −1.2 |
| DMT003052 | 8.8±0.3 | 1.5 | −1.8 |
| DMT003102 | 14±2 | 1.5 | −1.9 |
| DMT003104 | 32±2 | 12.3 | 1.3 |
| DMT003106 | 7.1±0.4 | 1.0 | −1.1 |
| DMT003108 | 7.5±0.8 | −1.1 | −1.1 |
| DMT003110 | >50 | 10.3 | −1.3 |
| DMT003020 | >50 | 1.1 | −2.2 |
| DMT003024 | >50 | 2.3 | −5.0 |
| PTX(nM) | 6.1±1.2 | N/A | N/A |
Table 3.
Effects of select compounds on the growth of MCF7 breast cancer cells and HT29 colon cancer cells. The GI50 for the indicated compounds (in µM) was obtained as described in the Materials and Methods.
| MCF7 GI50 | HT29 GI50 | |
|---|---|---|
| MAL3-101 | 14.7±0.7 | >50 |
| MAL3-51 | >50 | >50 |
| DMT003088 | 2.4±0.8 | 4.8±0.3 |
| DMT003132 | 6.3±0.2 | 5.3±2 |
| DMT003052 | 7.8±0.3 | 3.1±0.4 |
The majority of the compounds with effects on cell growth had improved physicochemical attributes relative to MAL3-101, including reduced molecular weight, increased solubility, and decreased lipophilicity. For example, the predicted water solubility (logS) of MAL3-101 is −11.7 and the predicted lipophilicity (clogP) is 10.3 (QikProp, version 2.1). By comparison, for DMT003086 these values improved to −10.9 and 8.0, respectively, and for DMT003132 even greater improvements to −9.7 and 7.8, respectively, were noted.
Interestingly, inhibition of breast cancer growth was limited to two classes of MAL3-101 derivatives, Class 3 and 5. Members of these classes are pyrimidinone-peptoid hybrid molecules that have variations in either the aldehyde-derived group of the dihydropyrimidinone or the amine group of the peptoid strand. The difference between molecules in Class 1 and 3 is the presence of the peptoid component, which suggests that this segment is essential for inhibiting cell proliferation. Compounds in Class 1 are acids and probably cross the cell membrane inefficiently. It is possible that the presence of the peptoid increases lipophilicity and subsequent entry into the cell. However, the presence of the peptoid component alone is insufficient for anti-proliferative activity, as compounds in Class 4, which contain a peptoid, had no effect on breast cancer cell growth (Table 2). In addition, our results indicate that specific aldehyde- or amine-derived substituents differentially affect breast cancer cell proliferation. For example, compounds in Class 4 had various substituents at R2 in place of the 5-formyl-2-methoxycarbonylmethoxybenzoic acid methyl ester present in MAL3-101. Replacement of the 5-formyl-2-methoxycarbonylmethoxybenzoic acid methyl ester aldehyde utilized in the synthesis of MAL3-101 with any one of 10 distinct aldehydes resulted in compounds devoid of anti-proliferative activity. In contrast, GI50 values were sensitive to variations in the aldehyde-derived component of the pyrimidinone (Class 3) or the amine (Class 5) of the peptoid. This suggests that the p-biphenylcarboxaldehyde and n-hexylamine groups found in MAL3-101 are not critical for inhibition of breast cancer cell growth. Indeed, certain modification of these groups increased anti-proliferative activity significantly. For example, DMT003086 (Class 3) inhibited breast cancer proliferation with a GI50 of 6.0 µM and DMT003106 (Class 5) had a GI50 of 7.1 µM (Table 1).
Based on the known impact of Hsp70 function on breast cancer cell survival (see above), each compound was then examined for its effects on the single turnover ATPase activity of Hsp70. The single turnover assay utilizes Hsp70 that is pre-bound to ATP, thus ensuring that the rate-limiting ATP hydrolytic step (KCAT) is monitored independent of ATP binding and ATP release. For these assays, we employed the yeast Hsp70, Ssa1p, which can be purified in high quantities with few contaminants39. We also examined the effects of the compounds on the ability of a model J domain-containing protein, the SV40 TAg, to activate Hsp70’s ATPase activity. TAg can be purified in large quantities and selective inhibitors of TAg may be useful against polyomavirus infections. Furthermore, we previously showed that TAg and endogenous yeast J domain-containing proteins stimulate Ssa1p to a similar extent29.
As shown in Table 2 and in Figure 2, the chaperone modulators fell into several groups, and in some but not all cases their attributes correlated with the chemical classifications presented in Table 1. Interestingly, many compounds inhibited J domain stimulation of Hsp70 but had no effect on the endogenous ATPase activity of Hsp70. For example, in Class 1, DMT003036 had no effect on Hsp70 ATP hydrolysis, but inhibited Tag-stimulated ATP hydrolysis by 3.5 fold (Table 2, Figure 2A). Some molecules in Class 3 and 5, which inhibited breast cancer cell growth, had similar biochemical activities: DMT003084, in Class 3, inhibited TAg stimulation of Hsp70 by 5.2 fold (Table 2, Figure 2B), and DMT003102, in Class 5, inhibited TAg stimulation by 1.9 fold (Table 2, Figure 2D).
Figure 2.
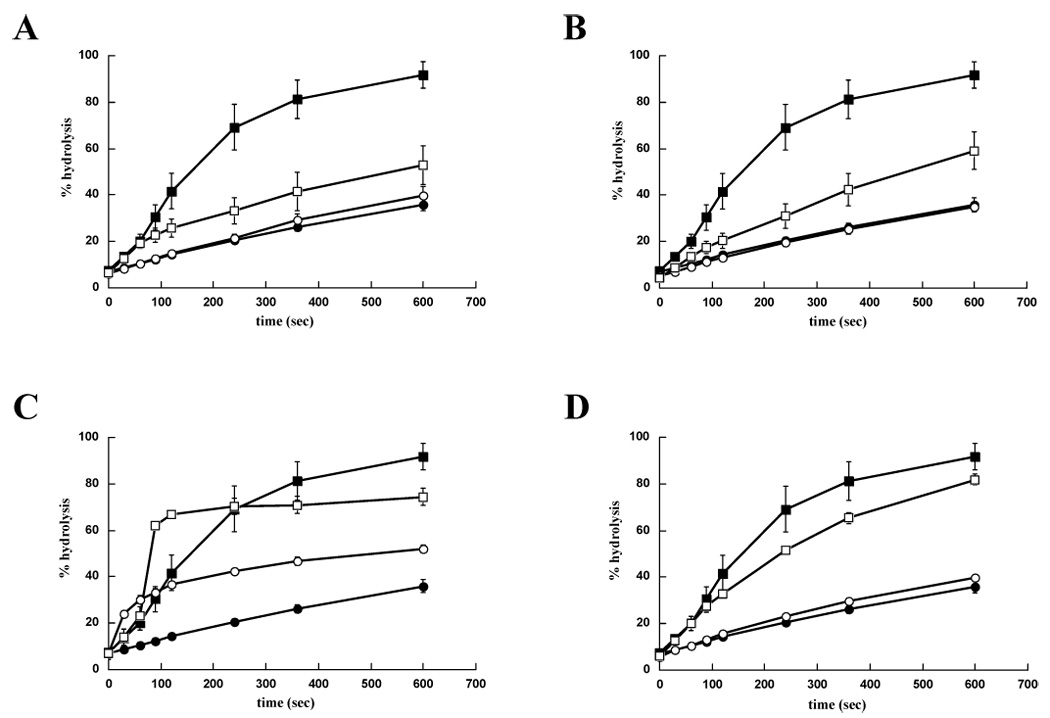
The MAL3-101 derivatives have distinct effects on chaperone activity. The endogenous and TAg-stimulated ATPase activities of Hsp70 for prototypical members of (A) Class 1 (e.g.DMT003036) (B) Class 3 (e.g. DMT003084) (C) Class 4 (e.g. DMT002220) (D) Class 5 (e.g. DMT003102) are shown. Filled circles, DMSO; filled squares 0.4 µM TAg and DMSO; open circles, 300 µM of the indicated compound; open squares, 0.4 µM TAg and 300 µM of the indicated compound.
Despite being able to inhibit the TAg stimulation of Hsp70, compounds in Class 1 had no effect on breast cancer cell proliferation, as exemplified by DMT003036 (Table 2). Since the only difference between members of Class 1 and Class 3, many of which inhibit SK-BR-3 proliferation, is the presence of the peptoid component, this suggests that the pyrimidinone is sufficient for inhibition of J protein stimulation of Hsp70. In contrast, the peptoid component is required for anti-proliferative activity, potentially by altering the lipophilicity and cell permeability of the molecules, but is insufficient on its own to confer this activity (also see above). In addition, the aldehyde-derived substituent attached to the pyrimidinone affects the biochemical and anti-proliferative activity of the compound. For example, the presence of the p-tert-butylphenyl group in either DMT003036 (Class 1) or DMT003084 (Class 3) greatly enhanced co-chaperone stimulation of Hsp70 (Figure 2A–B, Table 1–2), whereas the p-nitrophenyl group in both MAL2-13 and DMT003092 (Table 1–2) had little effect. Moreover, the magnitude of the anti-proliferative activity was sensitive to the 4-substituents in the dihydropyrimidinones. The compounds with phenyl (DMT003088) or p-nitrophenyl (DMT003086 and DMT003092) groups at this position were among the strongest inhibitors of breast cancer cell proliferation (Table 2). Also, the addition of aliphatic groups derived from cyclohexylcarboxaldehyde (DMT003100) and cyclopropylcarboxaldehyde (DMT003132) was important for the inhibition of breast cancer cell proliferation. These results suggest that the future development of a structure activity relationship for Hsp70 modulation and reduced breast cancer cell proliferation is feasible.
It is interesting to note that inhibition of breast cancer cell proliferation was not always directly related to modulation of co-chaperone activity. For example, DMT003088 and DMT003092 were two of the strongest inhibitors of breast cancer cell growth, but had little effect on TAg stimulation of Hsp70 (Table 2). There are several explanations for these data: First, these compounds might differentially affect the properties of distinct J domain-containing proteins, only one of which may interact with Hsp70 and promote cell survival. In fact, MAL3-101 exerts different effects on the abilities of TAg and the yeast J domain-containing protein, Ydj1p, to enhance Hsp70 ATPase activity29. Second, the compounds might be metabolized within the cell to generate anti-proliferative compounds. Third, the compound’s anti-proliferative effect might be through an alternate mechanism. Conversely, some compounds that had strong effects on chaperone function had little impact on SK-BR-3 proliferation. Here too, several scenarios can be envisaged: First, the compounds may not be cell permeable. While this may be likely for the acids in Class 1, it is an unlikely explanation for the inactive pyrimidinone-peptoid hybrids in Class 3 and 5 since they have similar solubilities and lipophilicities as the active compounds (e.g., DMT003086 and DMT003106). Second, compounds that are cell permeable could be metabolized once inside the cell. Finally, the TAg/Ssa1p chaperone interaction may not be the most appropriate indicator for anti-proliferative effects on SK-BR-3 growth.
Our data also indicate the existence of new classes of Hsp70 modulators. For example, the pyrimidinone molecules containing a methyl ester (Class 2) moderately stimulated Ssa1p ATP hydrolysis, but had a limited effect on TAg induced ATP hydrolysis in single turnover ATPase assays (data not shown). Some compounds in Class 4 represent another new class of chaperone modulator. For example, DMT002220 dramatically stimulated both Ssa1p ATP hydrolysis and TAg stimulated Ssa1p ATP hydrolysis (Figure 2C, Table 2). Although compounds in Class 2 and 4 had no effect on breast cancer cell proliferation, they may serve as modifiers for protein conformational diseases: The Gestwicki lab recently described an Hsp70 agonist which inhibited the in vitro aggregation of the β amyloid protein linked to Alzheimer’s disease40.
In conclusion, we have identified several classes of Hsp70 modulators that inhibit cell proliferation. Based on the fact that Hsp70 exhibits a strong anti-apoptotic effect at several distinct nodes in the apoptotic pathway14–16, we believe that the inhibitors of cell proliferation act directly on Hsp70 and induce pre-apoptotic phenomena. This, in turn, slows and ultimately arrests cell growth. This model is consistent with the fact that GI50 for MAL3-101 in SK-BR-3 and MCF7 cells reported here (14–27 µM) matches well with the ED50 for this compound to induce apoptosis in SK-BR-3 cells33 (~8 µM). Many of these compounds also have improved physicochemical characteristics, such as decreased molecular weight and lipophilicity and increased solubility. Continued efforts will seek to improve the potency of these compounds and to examine their effects in other cellular models for human diseases.
3. Experimental Methods
3.1 General method for the synthesis of compounds in classes 1–5
The synthesis of MAL3-101 was previously reported29 employing successive Biginelli41 and Ugi reactions to create a pyrimidinone-peptoid hybrid (Figure 1). In brief, benzyl acetoacetate and 6-ureidohexanoic acid were combined with p-biphenylcarboxaldehyde and reacted in a solution of tetrahydrofuran (THF) and HCl. The product, MAL2-11B, was combined with n-butylisocyanide, 5-formyl-2-methoxycarbonylmethoxybenzoic acid methyl ester, and n-hexylamine in an Ugi condensation reaction to create MAL3-101. The MAL3-101 derivatives were synthesized in similar reactions, but one reactant was altered to create an indexed library34. The purities of the compounds were determined using a variety of methods (evaporative light scattering, total ion count/MS, and UV spectroscopy) and the data for each compound are shown in Supplemental Table 1.
3.1.1 Synthesis of Class 1 pyrimidinones
Compounds in this class were synthesized as described previously34 and contain only the heterocyclic pyrimidinone product of the Biginelli reaction. The aldehyde component utilized in each reaction is shown in Table 1.
3.1.2 Synthesis of methyl 6-ureidohexanoate (Class 2)
A solution of 6-ureidohexanoic acid (0.996 g, 5.72 mmol, 1.0 eq) in methanol/benzene (2/7, 60 mL) was cooled to 0 °C, and a solution of trimethylsilyldiazomethane in hexanes (2.0 M, 3.43 mL, 6.86 mmol, 1.2 eq) was added. The reaction mixture was warmed to room temperature and stirred for 1 h prior to removing all volatile components in vacuo. The crude methyl ester (1.06 g, 5.61 mmol, 98%) was used without further purification in the Biginelli reaction as previously described34 (Scheme 1).
3.1.3. Synthesis of pyrimidinone-peptoid hybrids (Classes 3–5)
The synthesis of these compounds was previously reported34. Class 3 compounds utilize the compounds synthesized in Class 1 as the pyrimidinone building block, which is further reacted in the Ugi multicomponent condensation. Compounds in Class 4 and 5 utilize p-biphenylcarboxaldehyde as the aldehyde reagent in the Biginelli reaction and an assortment of aldehydes (Class 4) or esters (Class 5) in the Ugi reaction.
3.2. Synthesis of the MAL3-101 diacid (Class 6) and DMT003024 (Class 7)
The MAL3-101 diacid was synthesized by reacting MAL3-101 in the presence of NaOH as described previously34. DMT003024 (benzyl 1-(6-((1-(biphenyl-4-yl)-2-(butylamino)-2-oxoethyl)(2-morpholinoethyl)amino)-6-oxohexyl)-6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate) was prepared as follows: 4-Phenylbenzylaldehyde (1.0 eq) and 4-(2-aminoethyl)morpholine (1.0 eq) were added at room temperature to a suspension of dihydropyrimidinone (1.0 eq) in methanol. After stirring the reaction mixture for 10 min, n-butylisocyanide (1.0 eq) was added and the reaction mixture was heated to reflux for 24 h. After cooling to room temperature and concentration in vacuo, the residue was extracted with EtOAc, washed twice with an aqueous 10% NaOH solution, once with brine, and dried (MgSO4) prior to purification by chromatography on SiO2 on an ISCO Companion chromatography system. The product was obtained as a 2:1 mixture of diastereomers based on 1H NMR integration of triplets at δ 0.88 and 0.94 ppm, respectively. Major diasteromer: 1H NMR (300 MHz, CDCl3) δ 8.06 (d, 2 H, J = 8.7 Hz), 7.62-7.25 (m, 14 H), 7.17-7.14 (m, 2 H), 6.56 (bs, 0. 8H), 5.90 (d, 0.4 H, J = 2.3 Hz), 5.44 (bs, 0.9 H), 5.13 (d, 1H, J = 12.2 Hz), 5.01 (d, 1 H, J = 12.2 Hz), 3.83-3.16 (m, 10 H), 2.54 (s, 3 H), 2.44-1.99 (m, 9 H), 1.69-1.25 (m, 10 H), 0.88 (t, 2 H, J= 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 173.6, 169.6, 165.3, 153.0, 150.3, 147.3, 141.3, 140.1, 135.5, 134.3, 133.2, 129.8, 129.4, 128.8, 128.4, 128.2, 127.6, 127.3, 127.2, 126.9, 123.8, 102.7, 66.5, 66.2, 53.5, 53.3, 39.3, 33.1, 31.3, 29.6, 26.4, 24.8, 19.9, 16.2, 13.6; MS (ESI) m/z (rel intensity) 859 ([M+H]+, 100); HRMS (ESI) m/z calcd for C49H59N6O8 859.4316, found 859.4377. Characteristic signals of minor diasteromer: 1H NMR (300 MHz, CDCl3) δ 8.80 (bs, 0.2 H), 6.27 (m, 0.4 H), 5.53 (bs, 0.3 H), 0.94 (t, 1 H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 174.3, 170.1, 65.3, 62.4, 57.6, 56.8, 43.7, 39.5, 33.8, 32.1, 26.1, 20.2, 13.7.
3.5 Synthesis of N-(3-((tert-Butylcarbamoyl-(4-iodophenyl)methyl)-(2-morpholin-4-ylethyl)carbamoylpropylbenzamide (MAL3-51)
According to the general protocol B reported in ref. 29, 4-iodobenzaldehyde (0.982 g, 4.23 mmol), 4-(2-aminoethyl)morpholine (0.547 g, 0.552 mL, 0.423 mmol), 4-benzoylaminobutyric acid (0.877 g, 0.423 mmol) and tert-butylisocyanide (0.349 g, 0.474 µL, 0.423 mmol) in MeOH (10 mL) afforded MAL3-51 (2.02 g, 3.19 mmol, 75%) as colorless crystals after crystallization from ethyl acetate/hexanes (1/1): Mp 109 °C; IR (film) 3261, 2966, 1712, 1677, 1552, 1119 cm−1; 1H NMR (300.1 MHz, CDCl3) δ 82 (d, 2 H, J = 8.3 Hz), 7.65 (d, 2 H, J = 8.3 Hz), 7.51-7.39 (m, 3 H), 7.25 (t, 1 H, J = 4.8 Hz), 7.04 (d, 2 H, J = 6.1 Hz), 5.79 (s, 1 H), 5.68 (s, 1 H), 3.65-3.31 (m, 8 H), 2.64-1.81 (m, 10 H), 1.31 (s, 9 H); 13C NMR (CDCl3) δ 180.2, 174.2, 173.1, 141.8, 138.9, 137.9, 134.7, 131.7, 130.1, 95.4, 66.0, 61.8, 56.3, 52.1, 50.1, 42.0, 37.3, 28.0, 25.5, 20.9; MS (EI) m/z (rel. intensity) 634 (M+, 10), 522 (10), 113 (40), 100 (100); HRMS (EI) m/z calculated for C29H39IN4O4 634.2016, found 634.2022.
3.4 Cell Proliferation Assay
The cell proliferation assays were performed as previously described 35, 36 using the breast cancer cell lines, SK-BR-3 and MCF-7, and the colon cancer cell line, HT29, where indicated. In brief, cells were maintained as exponentially growing cultures in DMEM medium with 10% FBS, 1% penicillin, and 1% glutamine. The cells were seeded (4,000 cells per well) into 96-well plates and allowed to attach and grow for 48 h. One plate of cells was used for a time zero cell number determination (N = 16), and cells in other plates were treated for 72 h with either DMSO (1 v/v; N = 8 for each plate) or a range of concentrations, in quadruplicate, of test agents. Cell number was determined spectrophotometrically at 490 nm minus the absorbance at 630 nm (background absorbance) after exposure to 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium (MTS) and N-methylphenazine methylsulfate. The 50% growth inhibitory concentrations (GI50) of test agents were calculated over the 72 h or 96 h period. If the concentration required to obtain a GI50 was higher than 50 µM, this is reported as >50 in Table 2.
3.5. Hsp70 Single Turnover ATPase Assays
Yeast Hsp70 (Ssa1p) and SV40 Tag were purified as described previously39, 42 and single turnover ATPase assays were performed as published29. Endogenous Hsp70 ATPase assays had 300 µM test compound or DMSO added at 0 sec, whereas the TAg stimulation experiments had the compounds added at 60 sec. For each experiment, the timepoint at which compound was added was set to zero, results from multiple experiments were averaged, and kinetic data were obtained using KaleidaGraph. The results were fit to a single exponential using the equation: % Product= A × (1-exp(-kt)), where A is the amplitude of the reaction, k is the rate of ATP hydrolysis and t is time. The slope of the line for each compound was compared to the slope of the DMSO control and is reported as the fold change. All reported data represent the means of two or more independent reactions and error bars represent standard deviations of the data.
Supplementary Material
Acknowledgements
The authors would like to thank Paul Cantalupo for purified TAg and Susan Gilbert for valuable discussions, and Claire Coleman and Raghavan Balachandran for data analysis and technical assistance. This work was supported by a Partnership for Cures and Goldman Philanthropic Partnerships grant to J. L. B. C.M.W. was supported by a Renal Epithelial Biology training grant (DK61296) and a Mellon Graduate Fellowship, and R. J. C. was supported by a fellowship from the Beckman Foundation. P.W., D.M.T., S.W., and D.M.H. thank the NIH-NIGMS for grant P50-GM067082 for support of the CMLD program at the University of Pittsburgh.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mayer MP, Bukau B. Cell Mol Life Sci. 2005;62:670. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bukau B, Weissman J, Horwich A. Cell. 2006;125:443. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Fewell SW, Travers KJ, Weissman JS, Brodsky JL. Annu Rev Genet. 2001;35:149. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KM, DeLuca-Flaherty C, McKay DB. Nature. 1990;346:623. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 5.Wang TF, Chang JH, Wang C. J Biol Chem. 1993;268:26049. [PubMed] [Google Scholar]
- 6.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Science. 1996;272:1606. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craig EA, Huang P, Aron R, Andrew A. Rev Physiol Biochem Pharmacol. 2006;156:1. doi: 10.1007/s10254-005-0001-0. [DOI] [PubMed] [Google Scholar]
- 8.Greene MK, Maskos K, Landry SJ. Proc Natl Acad Sci U S A. 1998;95:6108. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wall D, Zylicz M, Georgopoulos C. J Biol Chem. 1994;269:5446. [PubMed] [Google Scholar]
- 10.Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y, Whyte P, Rundell K, Brodsky JL, Pipas JM. Mol Cell Biol. 1997;17:4761. doi: 10.1128/mcb.17.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allander T, Andreasson K, Gupta S, Bjerkner A, Bogdanovic G, Persson MA, Dalianis T, Ramqvist T, Andersson B. J Virol. 2007 doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eash S, Manley K, Gasparovic M, Querbes W, Atwood WJ. Cell Mol Life Sci. 2006;63:865. doi: 10.1007/s00018-005-5454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jolly C, Morimoto RI. J Natl Cancer Inst. 2000;92:1564. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- 15.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Trends Biochem Sci. 2006;31:164. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Ciocca DR, Calderwood SK. Cell Stress Chaperones. 2005;10:86. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volloch VZ, Sherman MY. Oncogene. 1999;18:3648. doi: 10.1038/sj.onc.1202525. [DOI] [PubMed] [Google Scholar]
- 18.Seo JS, Park YM, Kim JI, Shim EH, Kim CW, Jang JJ, Kim SH, Lee WH. Biochem Biophys Res Commun. 1996;218:582. doi: 10.1006/bbrc.1996.0103. [DOI] [PubMed] [Google Scholar]
- 19.Melendez K, Wallen ES, Edwards BS, Mobarak CD, Bear DG, Moseley PL. Cell Stress Chaperones. 2006;11:334. doi: 10.1379/CSC-187.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciocca DR, Clark GM, Tandon AK, Fuqua SA, Welch WJ, McGuire WL. J Natl Cancer Inst. 1993;85:570. doi: 10.1093/jnci/85.7.570. [DOI] [PubMed] [Google Scholar]
- 21.Barnes JA, Dix DJ, Collins BW, Luft C, Allen JW. Cell Stress Chaperones. 2001;6:316. doi: 10.1379/1466-1268(2001)006<0316:eoihet>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Proc Natl Acad Sci U S A. 2000;97:7871. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadler SG, Tepper MA, Schacter B, Mazzucco CE. Science. 1992;258:484. doi: 10.1126/science.1411548. [DOI] [PubMed] [Google Scholar]
- 24.Tanabe K, Tokumoto T, Ishikawa N, Shimizu T, Okuda H, Ito S, Shimmura H, Inui M, Harano M, Ohtsubo S, Manu M, Shiroyanagi Y, Yagisawa T, Fuchinoue S, Toma H. Transplant Proc. 2000;32:1745. doi: 10.1016/s0041-1345(00)01427-5. [DOI] [PubMed] [Google Scholar]
- 25.Ishida H, Miyamoto N, Shirakawa H, Shimizu T, Tokumoto T, Ishikawa N, Shimmura H, Setoguchi K, Toki D, Iida S, Teraoka S, Takahashi K, Toma H, Yamaguchi Y, Tanabe K. Am J Transplant. 2007 doi: 10.1111/j.1600-6143.2006.01676.x. [DOI] [PubMed] [Google Scholar]
- 26.Nadeau K, Nadler SG, Saulnier M, Tepper MA, Walsh CT. Biochemistry. 1994;33:2561. doi: 10.1021/bi00175a027. [DOI] [PubMed] [Google Scholar]
- 27.Brodsky JL. Biochem Pharmacol. 1999;57:877. doi: 10.1016/s0006-2952(98)00376-1. [DOI] [PubMed] [Google Scholar]
- 28.Nadler SG, Dischino DD, Malacko AR, Cleaveland JS, Fujihara SM, Marquardt H. Biochem Biophys Res Commun. 1998;253:176. doi: 10.1006/bbrc.1998.9775. [DOI] [PubMed] [Google Scholar]
- 29.Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. J Biol Chem. 2004;279:51131. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- 30.Fewell SW, Day BW, Brodsky JL. J Biol Chem. 2001;276:910. doi: 10.1074/jbc.M008535200. [DOI] [PubMed] [Google Scholar]
- 31.Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. EMBO Rep. 2004;5:567. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennessy F, Nicoll WS, Zimmermann R, Cheetham ME, Blatch GL. Protein Sci. 2005;14:1697. doi: 10.1110/ps.051406805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y, Wu N, Felts S, Wipf P, Massague J, Jiang X, Brodsky JL, Krystal GW, Chiosis G. Nat Chem Biol. 2007;3:498. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- 34.Werner S, Turner DM, Lyon MA, Huryn DM, Wipf P. SYNLETT. 2006;14:2334. [Google Scholar]
- 35.Minguez JM, Kim SY, Giuliano KA, Balachandran R, Madiraju C, Day BW, Curran DP. Bioorg Med Chem. 2003;11:3335. doi: 10.1016/s0968-0896(03)00186-x. [DOI] [PubMed] [Google Scholar]
- 36.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 37.Bhalla KN. Oncogene. 2003;22:9075. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- 38.Wang TH, Wang HS, Soong YK. Cancer. 2000;88:2619. doi: 10.1002/1097-0142(20000601)88:11<2619::aid-cncr26>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 39.McClellan AJ, Brodsky JL. Genetics. 2000;156:501. doi: 10.1093/genetics/156.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans CG, Wisen S, Gestwicki JE. J Biol Chem. 2006;281:33182. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 41.Wipf P. Cunningham April. Tetrahedron Lett. 1995;36:7819. [Google Scholar]
- 42.Cantalupo P, Saenz-Robles MT, Pipas JM. Methods Enzymol. 1999;306:297. doi: 10.1016/s0076-6879(99)06019-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.