

Crystal structure analysis of the apo form of catabolite control protein A reveals the three-helix bundle of the DNA-binding domain. In the crystal packing, this domain interacts with the binding site for the corepressor protein.
Keywords: catabolite control protein A, carbon catabolite repression, Bacillus megaterium
Abstract
Crystal structure determination of catabolite control protein A (CcpA) at 2.6 Å resolution reveals for the first time the structure of a full-length apo-form LacI-GalR family repressor protein. In the crystal structures of these transcription regulators, the three-helix bundle of the DNA-binding domain has only been observed in cognate DNA complexes; it has not been observed in other crystal structures owing to its mobility. In the crystal packing of apo-CcpA, the protein–protein contacts between the N-terminal three-helix bundle and the core domain consisted of interactions between the homodimers that were similar to those between the corepressor protein HPr and the CcpA N-subdomain in the ternary DNA complex. In contrast to the DNA complex, the apo-CcpA structure reveals large subdomain movements in the core, resulting in a complete loss of contacts between the N-subdomains of the homodimer.
1. Introduction
In Gram-positive bacteria, carbon catabolite repression (CCR) is regulated at the level of transcription by catabolite control protein A (CcpA), which controls the catabolic genes. In bacilli and other Gram-positive bacteria with low GC content, CcpA is the master transcriptional regulator; for example, approximately 10% of the Bacillus subtilis genome is under regulation by CcpA (Moreno et al., 2001 ▶). This regulator has both activator and repressor activities. Its binding to the catabolite responsive element (cre) DNA sites is regulated by phosphorylation of the histidine-containing proteins HPr or Crh. Several studies have demonstrated that the Ser46-phosphorylated form of HPr binds CcpA as a corepressor, activating binding to cre sites (Galinier et al., 1999 ▶; Aung-Hilbrich et al., 2002 ▶). Recently, a comparison of the crystal structures of the ternary complexes CcpA–(HPr-Ser46-P)–DNA and CcpA–(Crh-Ser46-P)–DNA with structures of the CcpA core domain provided a structural basis for the allosteric mechanisms activating CcpA binding to the cre sites (Schumacher et al., 2004 ▶, 2006 ▶). Other crystal structures of members of the LacI-GalR family bound to cognate DNA have been reported for the purine repressor PurR (Schumacher et al., 1994 ▶) and the lactose repressor LacI (Lewis et al., 1996 ▶).
Like PurR and LacI, the quaternary structure of CcpA is a homodimer. The monomer consists of an N-terminal DNA-binding domain and a larger C-terminal effector or corepressor-binding core domain which is also responsible for dimerization. The DNA-binding domain is a helix–turn–helix motif containing a three-helix bundle which is attached to the core domain by a short minor-groove-binding hinge helix. The large C-terminal core domain, similar to the periplasmic sugar-binding proteins (Quiocho & Ledvina, 1996 ▶), is divided into N- and C-subdomains connected by a crossover region. The relative orientation of the subdomains determines the DNA-binding ability. This is regulated by the presence or absence of small effector molecules for PurR and LacI, but in the case of CcpA the specific allosteric effectors are phosphorylated proteins (Schumacher et al., 2004 ▶, 2006 ▶).
Crystal structure analyses of DNA complexes of these regulator proteins revealed the full-length polypeptides in the electron-density maps. In all other crystal structures, the N-terminal DNA-binding domain (the first 60 amino-acid residues) was proteolysed to support crystallization or could not be observed in the electron-density maps owing to its high mobility. In contrast, here we report the crystal structure of the apo form of CcpA from B. megaterium as the first full-length structure of a LacI-GalR member which is not bound to DNA.
2. Materials and methods
The recombinant protein was expressed in Escherichia coli and was purified as described by Deutscher et al. (1995 ▶). Crystallization, X-ray diffraction data collection under cryoconditions and data processing have been described previously (Tebbe et al., 2000 ▶). The protein with an N-terminal His6 tag of 17 amino-acid residues (MRGSHHHHHHGSDDDDK) was crystallized using the hanging-drop vapour-diffusion method. CcpA solution (2.5 µl) containing 20 mg ml−1 protein in 10 mM Tris–HCl buffer pH 7.5 and 5 mM glucose-6-phosphate was mixed with the same volume of reservoir solution and allowed to equilibrate against 1 ml reservoir solution [3% polyethylene glycol 8000, 100 mM CaCl2, 10%(v/v) glycerol in 100 mM Tris–HCl buffer pH 8.0]. Hexagonal bipyramidal crystals grew within several days and were transferred to reservoir solution containing 25% glycerol as cryoprotectant prior to data collection. X-ray diffraction data from flash-frozen crystals at 100 K were measured at beamline X11 (EMBL Outstation, Hamburg) and processed using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶).
The structure of CcpA was solved by molecular replacement using AMoRe from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The search model was a monomer of the proteolysed core domain (residues 60–332) of CcpA from B. megaterium (PDB codes 1sxg, 1sxh and 1sxi) taken from Schumacher et al. (2004 ▶). Monomers of all three structures produced the same solution in the resolution range 15–4 Å. After cross-rotation, the translation results of AMoRe unambiguously distinguished between the enantiomorphic space groups P6122 and P6522. The resulting model along with the scaled intensities was submitted to the TB Structural Genomics Consortium bias-removal server (Reddy et al., 2003 ▶) to minimize the model bias. Initial refinement of the model was performed with simulated annealing using the CNS package (Brünger et al., 1998 ▶). This revealed the DNA-binding domain (Fig. 1 ▶), which was not part of the search model, in the first electron-density maps. Model building was performed using O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). The final refinement was performed with REFMAC5 (Murshudov et al., 1999 ▶) including as TLS groups the N-terminal three-helix bundle and the N- and C-subdomains. Structure alignments and superposition were performed with ALIGN (Cohen, 1997 ▶) and LSQKAB (Kabsch, 1976 ▶; Collaborative Computational Project, Number 4, 1994 ▶). Protein interfaces were calculated using the Protein Interfaces, Surfaces and Assemblies service PISA at the European Bioinformatics Institute (Krissinel & Hendrick, 2005 ▶). Figures were prepared with MOLSCRIPT (Kraulis, 1991 ▶), BOBSCRIPT (Esnouf, 1999 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶). The diffraction data statistics and the crystallographic refinement statistics are summarized in Table 1 ▶.
Figure 1.

The electron density shows an F o − F c OMIT map at the 2.5σ level calculated after refinement of the molecular-replacement solution based on the truncated apo-CcpA model. The DNA-binding domain was clearly identified and traced. The refinement of the truncated model converged with R = 0.317, R free = 0.371 and a figure of merit of 0.701. See Table 1 ▶ for final refinement statistics of the complete apo-CcpA. The side-chain orientations of Arg21 and Glu42 of the final model are shown along with a Cα trace.
Table 1. Data-collection and refinement statistics of apo-CcpA.
Values in parentheses are for the outer resolution shell.
| Data-collection statistics | |
| X-ray source | X11, EMBL Hamburg |
| Wavelength (Å) | 0.95 |
| Resolution limits (Å) | 99.0–2.55 (2.65–2.55) |
| Space group | P6122 |
| Unit-cell parameters (Å) | a = b = 74.45, c = 238.84 |
| Total reflections | 92728 |
| Unique reflections | 14161 |
| Redundancy | 6.5 |
| I/σ(I) | 24.8 (3.7) |
| Completeness (%) | 98.6 |
| Rsym† (%) | 5.1 (43.6) |
| B factor from Wilson plot (Å2) | 28.6 |
| Refinement statistics | |
| Resolution limits (Å) | 20–2.6 (2.7–2.6) |
| No. of reflections in working set/test set | 12815/1036 |
| Rcryst‡/Rfree§ (%) | 22.8/29.6 (28.9/33.1) |
| Figure of merit | 0.795 |
| R.m.s.d. bond lengths (Å) | 0.010 |
| R.m.s.d. angles (°) | 1.30 |
| Polypeptide chains in ASU | 1 |
| Matthews coefficient (Å3 Da−1) | 2.40 |
| No. of amino-acid residues | 322 |
| No. of protein atoms | 2469 |
| No. of solvent atoms | 42 |
| Average B factors (Å2) | |
| Main-chain atoms | 47.1 |
| Side-chain atoms | 47.7 |
| Solvent atoms | 51.6 |
| Ramachandran plot | |
| Favoured (%) | 88.7 |
| Allowed (%) | 10.3 |
| Disallowed | None |
R
sym = 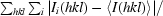
 , where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is the average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is the average.
R
cryst = 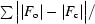
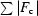 , where F
o and F
c are the observed and calculated structure-factor amplitudes, respectively.
, where F
o and F
c are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated using a randomly selected 8% of the data set that was omitted throughout all stages of refinement.
3. Results and discussion
The structure of apo-CcpA was solved at 2.6 Å resolution by molecular replacement using the C-terminal core domain as a search model. The asymmetric unit consists of a monomer, which forms the functional CcpA homodimer via a crystallographic twofold axis.
The polypeptide chain was successfully traced for the regulatory core domain and for the N-terminal DNA-binding domain, which was absent in the search model. This is the first report of a structure of the LacI-GalR family in which the DNA-binding domain is observed in the apo form. In all crystal structures of these repressor proteins, the DNA-binding domains are mobile in the absence of operator DNA, resulting in a complete lack of electron density for the three-helix bundle and the adjacent hinge-helix. To date, crystal structures of apo-CcpA are only available for the proteolytically truncated core domain, which has been crystallized in three different space groups (Schumacher et al., 2004 ▶). The complete DNA-binding domain is observed in the F o − F c electron-density map for the hexagonal packing of apo-CcpA (Fig. 1 ▶) and is positioned by lattice contacts. Only for the His tag and residues 52–59 is no density observed. The polypeptide segment from residues 52 to 59 connects the DNA-binding domain to the large regulatory core domain and forms the short helix α4 upon effector and DNA binding. This helix is responsible for minor-groove recognition, in addition to the helix–turn–helix motif (α1, α2) which binds to the major groove.
Interestingly, the DNA-binding three-helix bundle (residues 3–45, Fig. 2 ▶) of apo-CcpA has an r.m.s. deviation of only 0.6 Å based on aligned Cα positions with respect to the DNA complex (PDB codes 1rzr and 1zvv). The stability and folding of the three-helix bundle in solution is verified by the NMR structures of the comparable PurR and LacI headpiece (Nagadoi et al., 1995 ▶; Slijper et al., 1996 ▶). The lack of electron density for this domain in other crystallized full-length LacI-GalR proteins can be explained by high mobility of the DNA-binding three-helix bundle, supported by the unfolded polypeptide of helix α4 (Schumacher et al., 2004 ▶). The different position of the three-helix bundle in apo-CcpA compared with the DNA complex demonstrates its high mobility (Fig. 3 ▶). At a first glance, the DNA-binding domains in apo-CcpA are positioned by contacts to the N-subdomain of the other polypeptide in the homodimer. A closer look at the crystal packing shows a similarity to the protein–protein contacts in the ternary complexes of CcpA with DNA and the corepressor phosphoproteins HPr or Crh (Schumacher et al., 2004 ▶, 2006 ▶). In apo-CcpA, the DNA-binding domain of another homodimer forms several hydrophobic (Leu38–Met88) and hydrophilic (Glu42–Arg303, Arg47–Asp296) contacts at the corepressor-binding site (Fig. 4 ▶). Mutagenic studies revealed that CcpA residues Tyr89 and Tyr295, Ala299 and Arg303 are essential for HPr-Ser46-P binding (Kraus et al., 1998 ▶), which covers about 700 Å2 of the N-subdomain. In the crystal packing of apo-CcpA, the area buried by the DNA headpiece for this contact is about 500 Å2.
Figure 2.

Superposition of the DNA-binding three-helix bundles of apo-CcpA (blue), CcpA–(HPr-Ser46-P)–DNA (cyan, PDB code 1rzr) and CcpA–(Crh-Ser46-P)–DNA (green, PDB code 1zvv). The helix–turn–helix motif begins at Thr4.
Figure 3.

Cα trace of superposed homodimers of apo-CcpA (blue) and CcpA–(HPr-Ser46-P)–DNA (red, PDB code 1rzr). Superposition was performed with respect to the C-terminal subdomains (Csub) of CcpA. The twofold axis is in the vertical direction. (a) clearly shows the loss of contact between the N-terminal subdomains (Nsub), which exhibit a more open conformation in apo-CcpA (blue) compared with the ternary complex. In (b) CcpA is rotated around the dimer axis by about 90° to show the differences in the orientation of the DNA-binding domains (Dbd). Note also the cleft between the N- and C-subdomains, which is the small-molecule effector-binding site of the LacI-GalR family.
Figure 4.

The monomers of apo-CcpA and its DNA complex (PDB code 1rzr) are shown based on superposition of the N-subdomains (Nsub). In the apo-CcpA structure (dark colours), contacts are made to a symmetry-related mate of the DNA-binding domain (Dbd*, orange) at the same area where the corepressor Hpr binds in the DNA complex (light colours). The residues of the N-subdomain involved and discussed in the text are shown in ball-and-stick representation (blue). Also note the major movement of the DNA-binding domain by a rotation of 136°.
In the apo form, the large core domain of CcpA adopts a more open conformation compared with the homodimers of the DNA complex and the proteolysed core domain. Within the N- and C-subdomains the secondary-structure arrangements are almost unchanged, with r.m.s. deviations of Cα positions of only 0.86 and 0.6 Å, respectively. The striking difference appears upon superposition of the core domains of dimeric apo-CcpA and its DNA complex (PDB code 1rzr), showing an r.m.s. deviation of 6.1 Å based on aligned Cα positions (residues 60–332). Schumacher et al. (2004 ▶) compared the truncated CcpA with the DNA complex and described a β-strand ratcheting within the intersubunit β-sheet of the N-subdomains arising from a changed pattern of sheet-stabilizing hydrogen bonds at the twofold axis, which is near Leu95. In the apo-CcpA dimer this intersubunit β-sheet formation is not possible because the symmetry-related Cα positions of Leu95 are at a distance of 21.3 Å. In apo-CcpA, the N-subdomains completely lose their contacts with respect to the DNA-bound conformation and the truncated form (Fig. 3 ▶ a). The monomers of truncated CcpA have a contact surface of 1371 Å2, compared with apo-CcpA which exhibits a contact area of 708 Å2. These drastic conformational changes can be described by a rotation of the C-subdomains within the homodimer and a rotation of the N- to C-subdomain within each monomer. Compared with the DNA complex, the C-subdomains of apo-CcpA are rotated by about 24°. An additional rotation by 21° of the N- to C-subdomain is observed in each monomer. A comparison of the truncated CcpA (PDB code 1sxh) with the DNA complex (PDB code 1rzr) shows corresponding rotation values of only 8.5 and 4°, respectively. As a consequence of this, it might be difficult to explain the exact mechanism of conformational changes upon DNA binding of CcpA because packing effects of apo-CcpA and truncated CcpA cannot be ruled out. The newly discovered conformation of the quaternary structure of apo-CcpA makes previous reports on the mechanism of DNA binding questionable.
In the small-molecule-regulated repressors LacI and PurR, the cleft between the N- and C-subdomains (Fig. 3 ▶ b) is the binding site for effector molecules (Bell & Lewis, 2001 ▶; Schumacher et al., 1994 ▶). In this region of apo-CcpA, the truncated CcpA and the ternary CcpA–corepressor–DNA complexes show large deviations in a loop conformation N-terminal to α-helix 5 consisting of residues 70–73. Glucose-6-phosphate and fructose-1,6-diphosphate have been discussed as possible adjunct corepressor molecules that enhance CcpA binding to the effector protein HPr (Seidel et al., 2005 ▶). Glucose-6-phosphate was supplied in the crystallization buffer, but could not be observed in the electron-density maps. We therefore assume that only the protein–protein complex has sufficient affinity to bind glucose-6-phosphate.
Supplementary Material
PDB reference: CcpA, 2jcg, 2jcgsf
References
- Aung-Hilbrich, L. M., Seidel, G., Wagner, A. & Hillen, W. (2002). J. Mol. Biol.319, 77–85. [DOI] [PubMed] [Google Scholar]
- Bell, C. E. & Lewis, M. (2001). J. Mol. Biol.312, 921–926. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Cohen, G. E. (1997). J. Appl. Cryst.30, 1160–1161. [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Deutscher, J., Küster, E., Bergstedt, U., Charrier, V. & Hillen, W. (1995). Mol. Microbiol.15, 1049–1053. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M. (1999). Acta Cryst. D55, 938–940. [DOI] [PubMed] [Google Scholar]
- Galinier, A., Deutscher, J. & Martin-Verstraete, I. (1999). J. Mol. Biol.286, 307–314. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923. [Google Scholar]
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Kraus, A., Küster, E., Wagner, A., Hoffmann, K. & Hillen, W. (1998). Mol. Microbiol.30, 955–963. [DOI] [PubMed] [Google Scholar]
- Krissinel, E. & Hendrick, K. (2005). CompLife 2005, edited by M. R. Berthold, R. Glen, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 163–174. Berlin/Heidelberg: Springer-Verlag.
- Lewis, M., Chang, G., Horton, N. C., Kercher, M. A., Pace, H. C., Schumacher, M. A., Brennan, R. G. & Lu, P. (1996). Science, 271, 1247–1254. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Moreno, M. S., Schneider, B. L., Maile, R. R., Weyler, W. & Saier, M. H. Jr (2001). Mol. Microbiol.39, 1366–1381. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed] [Google Scholar]
- Nagadoi, A., Morikawa, S., Nakamura, H., Enari, M., Kobayashi, K., Yamamoto, H., Sampei, G., Mizobuchi, K., Schumacher, M. A., Brennan, R. G. & Nishimura, Y. (1995). Structure, 3, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Quiocho, F. A. & Ledvina, P. S. (1996). Mol. Microbiol.20, 17–25. [DOI] [PubMed] [Google Scholar]
- Reddy, V., Swanson, S. M., Segelke, B., Kantardjieff, K. A., Pacchettini, J. C. & Rupp, B. (2003). Acta Cryst. D59, 2200–2210. [DOI] [PubMed] [Google Scholar]
- Schumacher, M. A., Allen, G. S., Diel, M., Seidel, G., Hillen, W. & Brennan, R. G. (2004). Cell, 118, 731–741. [DOI] [PubMed] [Google Scholar]
- Schumacher, M. A., Choi, K. Y., Zalkin, H. & Brennan, R. G. (1994). Science, 266, 763–770. [DOI] [PubMed] [Google Scholar]
- Schumacher, M. A., Seidel, G., Hillen, W. & Brennan, R. G. (2006). J. Biol. Chem.281, 6793–6800. [DOI] [PubMed] [Google Scholar]
- Seidel, G., Diel, M., Fuchsbauer, N. & Hillen, W. (2005). FEBS Lett.272, 2566–2577. [DOI] [PubMed]
- Slijper, M., Bonvin, A. M., Boelens, R. & Kaptein R. (1996). J. Mol. Biol.259, 761–773. [DOI] [PubMed] [Google Scholar]
- Tebbe, J., Orth, P., Küster-Schöck, E., Hillen, W., Saenger, W. & Hinrichs, W. (2000). Acta Cryst. D56, 67–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CcpA, 2jcg, 2jcgsf