


Abstract
Homing endonucleases initiate nonreciprocal transfer of DNA segments containing their own genes and the flanking sequences by cleaving the recipient DNA. Bacteriophage T4 segB gene, which is located in a cluster of tRNA genes, encodes a protein of unknown function, homologous to homing endonucleases of the GIY-YIG family. We demonstrate that SegB protein is a site-specific endonuclease, which produces mostly 3′ 2-nt protruding ends at its DNA cleavage site. Analysis of SegB cleavage sites suggests that SegB recognizes a 27-bp sequence. It contains 11-bp conserved sequence, which corresponds to a conserved motif of tRNA TψC stem-loop, whereas the remainder of the recognition site is rather degenerate. T4-related phages T2L, RB1 and RB3 contain tRNA gene regions that are homologous to that of phage T4 but lack segB gene and several tRNA genes. In co-infections of phages T4 and T2L, segB gene is inherited with nearly 100% of efficiency. The preferred inheritance depends absolutely on the segB gene integrity and is accompanied by the loss of the T2L tRNA gene region markers. We suggest that SegB is a homing endonuclease that functions to ensure spreading of its own gene and the surrounding tRNA genes among T4-related phages.
INTRODUCTION
Homing endonucleases have been described in bacteria, archaebacteria and eukaryotes (1–3). Open reading frames (ORFs) of these nucleases are found within group I introns, intergenic regions and inteins. It is often observed that homologous locus of a closely related organism lacks ORF of the homing endonuclease. This is consistent with the idea that homing endonucleases are a recent evolutionary acquisition. A natural target for cleavage by a homing endonuclease is DNA of the homologous locus that lacks its cognate ORF. When DNA molecules containing both loci are present in the same cell, the homing endonuclease introduces typically a double-strand break into the DNA of the locus lacking the endonuclease ORF. The introduced break initiates the double-strand break repair of the cleaved molecule using DNA of the homologous molecule containing the endonuclease ORF as the donor. The double-strand break repair results in a nonreciprocal transfer of a DNA segment that contains the endonuclease ORF and the flanking regions from the donor into the recipient. Such type of gene conversion is referred to as homing, and the enzymes that initiate this process are called homing endonucleases (reviewed in 4–6).
Based on the presence of conserved motifs, homing endonucleases are classified into one of the four families: LAGLI-DADG, GIY-YIG, H-N-H and His-Cys. Bacteriophage T4 contains genes for homing endonucleases I-TevI, I-TevII, SegA, SegE, SegF and SegG that belong to the GIY-YIG family (7–12). In addition, phage T4 contains segB-D and mobA-E genes, whose putative products share homology with homing endonucleases of the GIY-YIG and H-N-H family, respectively (13,14).
In contrast to I-TevI ORF, which is located within the td intron, segA-G ORFs are situated in the intergenic regions (11–13). segE is in the hoc.1-uvsW intergenic region and a cleavage site of SegE endonuclease is in the hoc.1-uvsW intergenic region of phages RB30 and RB52 that lack segE. In mixed infections of phages T4 and RB30, SegE initiates homing of its own gene with almost 100% efficiency (15). This process requires the segE gene integrity, depends on the DNA homology, and is accompanied by co-conversion of the flanking sequences. SegF and SegG endonucleases initiate similar processes in co-infection of phages T4 and T2L (11,12). As known, crosses of phages T4 and T2L are characterized by phenomenon of partial exclusion (16). The phenomenon consists in dominance of genetic markers of T4 over T2L in the progeny of mixed infection of bacteria by these phages. A fraction of the majority of phage T2L genetic markers in the progeny is lowered from the expected 50% to 10–40%. In addition, some T2L markers at local sites of the genome are strongly excluded and contribute no >1–2% to the progeny. Mechanism of the local marker exclusion had been unclear, until it was shown that SegF and SegG endonucleases are responsible for the exclusion of T2L genes 56 and 32, respectively (11,12), which was previously discovered to occur in mixed infection of these phages (16).
The segB gene is located between tRNAIle and tRNAArg genes of T4 in the tRNA gene region (13,17) and encodes a protein of unknown function. In contrast to bacteria, where tRNA genes are favorite sites for insertion of mobile group I introns (6), the presence of homing endonuclease genes in a tRNA gene cluster of phages is rarely observed (17,18). In this work we show that the segB gene codes for a site-specific endonuclease. The SegB endonuclease recognizes a 27-bp degenerate sequence containing a conserved element of the tRNA gene. Positions of the cleavage points relative to the main recognition element vary and often occur immediately after CCAR sequence. The segB gene is lacking in genomes of T4-related phages RB1, RB3 and T2L. In phage crosses between T4 and T2L, endonuclease SegB initiates homing of its own gene and the flanking sequences that excludes the phage T2L tRNA gene cluster converting it into a T4 variant.
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides #1: 5′-tttgtcgACTCATACCGCACTGATTAATACC-3′ [72574-7259], #2: 5′-tttctgcaGGCCATATCTCAACCATATCC-3′ (70970–70990), #3: 5′-ttcatATGTTCTATTACACTT-3′ (71903–71918), #4: 5′-ttggatcCATTACACCAG-3′ (71231–71241), #15: 5′-tttaagcTTACCAGAACCCATATATCCATCATC-3′ (71815–71840), #16: 5′-tttaagcTTAAGTCACGTTTTGAAAATGATATTTTAC-3′ (71546–71575), #17: 5′-ATTATTAAAAAGAGCCCAAGATAAATATGG-3′ (71785–71814), #18: 5′-ACGATTTAGCTTCTTTTAAGCTAGCATC-3′ (71576–71603), #19: 5′-TTCGATTCTCATTATCCGCTCC-3′ (72292–72313), #20: 5′-ATTTGGTATCCCGCCCTGG-3′ (71170–71188), #21: 5′-TTAAAATTTCATTTTTCGACCTTTAAACCA-3′ (71253–71282) and #26: 5′-tttctgcaGTTAATCACAACCTTTACAGTATACCACTGACC-3′ (70453–70485) were derived from T4 DNA (GenBank accession no. 000866). Positions of the oligonucleotide sequences in the phage T4 sequence are presented in the parentheses, and the uncomplimentary nucleotides are in the lower case letters.
Oligonucleotides #5: 5′-ACCACCATCTGGCGATTATGAG-3′ (1025–1046), #6: 5′-TTGGATGTGTAGCTCAATGGC-3′ (1168–1188), #7: 5′-TGGTCGAGGCAGTAGGG-3′ (1308–1324), #8: 5′-GAGAGCCTTTGTTAATAATTGG-3′ (25–46), #9: 5′-AGAATGGTCAAATTGGTAAAGG-3′ (121–142), #10: 5′-TTTGCGGATATCGTATAATGG-3′ (207–227), #11: 5′-TTTGGCCCTGTAGCTGG-3′ (991–1007), #12: 5′-GGGGAGTTATCCCGTAGAGGTAGC-3′ (1079–1102), #13: 5′-ACGAGGCATAGCTCAGAAGG-3′ (1248–1267), #14: 5′-ACTCCGTGTAGCTCAGTTTG-3′ (293–313), #22: 5′-GAATGGCTATTGGTGGAAATCAAC-3′ (586–609), #23: 5′-CTAGCTTGAGTATGGCTAACATTTATAGC-3′ (926–954), #24: 5′GAAAGGTAATGTTTATTTAGTCGTTCATG-3′ (1345–1373) and #25: 5′-CTTTTAAAGCATCAATCTGTTCACG-3′ (1661–1685) were derived from T2 DNA (EMBL accession no. AJ880101).
DNA constructs and DNA sequencing
DNA cloning and PCR were carried out according to the standard techniques (19). Re-sequencing of phage T4 segB ORF revealed that its length is 666 bp (GenBank accession no. Z69338) but not 606 bp as was determined earlier (GenBank accession no. X03016). Plasmid pSBET-15b contains the 666-bp ORF, which encodes phage T4 SegB protein, fused with six histidine residues at its N-terminus. The plasmid was created by cloning a phage T4 687-bp DNA fragment, amplified by PCR with primers #3 and #4 and then cleaved with NdeI and BamHI, into the same restriction endonuclease sites of pET-15b DNA (Novagen).
The 1957 bp T2L tRNA gene region and 1071 bp RB1 tRNA gene region were PCR-amplified with primers #1 and #2 and inserted into SalI–PstI sites of pUC19 (20) yielding plasmids pUT2LsB and pURB1sB, respectively.
Plasmid pBSsegB▵ carries the phage T4 tRNA gene cluster, where a part of the T4 segB ORF (nucleotides 105–343) is deleted. The deletion was created by two subsequent DNA clonings. A 778-bp T4 DNA fragment, amplified with primers #1 and #15, was first inserted into SalI–HindIII sites of pBluescriptSK(−) (Stratagene). In the second step, a 604-bp T4 DNA fragment, amplified with primers #2 and #16, was inserted in HindIII–PstI sites of the above plasmid yielding pBSsegB▵ plasmid.
The structure of all cloned DNA fragments was confirmed by DNA sequencing carried out using DTCS kit and CEQ2000XL DNA Analysis System (Beckman Coulter).
The DNA structures of the tRNA gene regions of phages T4, T2L, RB1 and RB3 amplified with primers #1 and #26 were determined by direct sequencing of both strands of the corresponding PCR fragments and were submitted to the EMBL database (EMBL accession numbers for the T2L, RB1 and RB3 sequences AJ880101, AJ884577 and AJ884578, respectively).
Bacteriophages and Escherichia coli strains
Escherichia coli JM109 strain was used as a host for plasmid construction and for crossings between plasmids and phages (20). Escherichia coli B40 (SuI) and BE (Sup°) strains were used as hosts to prepare phage stocks and to cross phages. Escherichia coli B834 strain (F− hsdRB hsdMB met thi Sup°) was used to propagate phages with 5-hydroxymethylcytosine or cytosine DNA (21).
Bacteriophages T4B, T4290 (αgt am8 βgt am10), T4alc7 (g56amE51 g42amC87 denB NB5060 alc7), T4amB20 (g14am), T2L, RB1, RB2 and RB3 were kindly provided by Dr Valery Tanyashin [IBPM of the Russian Academy of Sciences (RAS)].
Bacteriophage B20ΔsegB containing a deletion of nucleotides 105–343 in the segB ORF was constructed as follows: ∼104 plaque-forming units (p.f.u.) of phage B20 were plated on E. coli JM109 cells containing the plasmid pBSsegB▵ at 37°C. During such plating, homologous recombination between the recipient phage genome and the plasmid generates with a low-frequency progeny that acquired the segB gene deletion. To identify the progeny, genomic DNAs of the progeny plaques were transferred onto a nylon HybondN+ membrane (GE Healthcare). The transferred DNA was denatured and hybridized with a 32P-labeled DNA probe complementary to the deletion region. The probe was produced by PCR with primers #17 and #18 in the presence of [α-32P]dATP (5000 Ci/mmol, Institute of Bioorganic Chemistry, RAS). Phages, which gave a negative signal in the Southern hybridization, were selected. The presence of deletion in the selected phages was confirmed by PCR with primers #19 and #20 (deletion yields 905 bp PCR product, whereas the wild-type DNA generates 1144-bp PCR product). One of the selected phages was designated as bacteriophage B20ΔsegB.
Overproduction and purification of phage T4 SegB endonuclease
Escherichia coli BL21(DE3)/pLysE cells (22) were transformed with the plasmid pSBET-15b and the transformants were grown on LB plates containing ampicillin (50 µg/ml) and chloramphenicol (34 µg/ml) at 37°C. About 100 of the freshly grown colonies were suspended in LB, and the suspension was used to inoculate 1 l of 2 × YT medium supplemented with ampicillin (100 µg/ml) and chloramphenicol (34 µg/ml). The culture was grown at 26°C to OD590 = 0.5 (∼2 × 108 cells/ml) and, at that point, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.25 mM. The growth was continued under the same conditions for additional 3.5 h. Cells were collected by centrifugation at 8000g for 10 min, frozen and stored at −70°C.
All purification steps were carried out at 4°C. During purification, SegB was identified in column fractions by SDS–PAGE followed by Coomassie R-250 staining and by the ability to cleave DNA at the major site located in the phage T2L tRNAAsn gene. Cells suspended in buffer A (15 mM К, Na-phosphate, pH 7.1, 0.5 mM EDTA, 10 mM 2-mercaptoethanol and 0.2 M NaCl) were disrupted in the presence of protease inhibitor cocktail ‘Complete’ (Roche Applied Science) in a Soniprep ultrasonic disintegrator (MSE, UK) at a frequency of 20 kHz nine times 30 s each with 1-min intervals for cooling. Cell debris were removed by centrifugation at 39 000g for 30 min. Clarified lysate was loaded onto a 5-ml phosphocellulose P11 (Whatman) column equilibrated with buffer A. The column was washed with 25 ml of the same buffer, and proteins were eluted with 50 ml of a linear gradient of NaCl from 0.2 to 1.5 M. SegB eluted from the column at 0.95 M NaCl.
Fractions containing SegB were pooled and dialyzed against buffer B (15 mM К, Na-phosphate, pH 7.1, 80 mM NaCl, 10 mM 2-mercaptoethanol and 50 mM imidazole, pH 7.1) for 16 h. After dialysis, the fraction was loaded onto a 1.5-ml Ni–NTA agarose (Qiagen) column that was equilibrated with the dialysis buffer. The column was then consecutively washed three times with 8 ml of buffer B containing increasing concentrations of imidazole (50 mM, 100 mM and 130 mM). To elute SegB, the column was washed with 9 ml of buffer B containing 0.25 M imidazole, pH 7.1.
The Ni–NTA agarose fraction of SegB was loaded onto a 10-ml hydroxyapatite HRPT column (Bio-Rad) equilibrated with buffer C (0.15 M К, Na-phosphate, pH 7.1, 50 mM NaCl and 10 mM 2-mercaptoethanol). The column was then washed with eight volumes of the equilibration buffer, and proteins were eluted with 80 ml of a linear gradient of K, Na-phosphate from 0.15 M to 0.4 M. SegB eluted at 0.31 M of the К, Na-phosphate buffer.
Hydroxyapatite fractions containing SegB peak were pooled and loaded onto a Mono-S HR 5/5 column (GE Healthcare) equilibrated with buffer A. The column was then washed with 10 ml of buffer A, and proteins were eluted with 15 ml of a linear gradient of NaCl from 0.2 M to 1 M in buffer A. SegB eluted at 0.72 M NaCl. The purified protein fraction was consecutively dialyzed against a 500-ml buffer containing 20 mM Tris–HCl, pH 7.5, 200 mM NaCl, 10 mM 2-mercaptoethanol, 0.1 mM EDTA and a 200 ml buffer containing 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM 2-mercaptoethanol, 0.1 mM EDTA and 50% (v/v) glycerol. The dialyzed enzyme fraction was stored at −20°C.
Protein concentration was determined by the Bradford method using BSA as standard (23).
Analysis of SegB endonuclease activity
To measure SegB endonuclease activity, a standard 20 µl mixture contained 10 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 10 mM DTT, 100 mM NaCl, 0.1 mg/ml BSA, 0.02% Triton X-100, 0.5 µg DNA of plasmid pURB1sB, linearized with restriction endonuclease Eco31I (or 0.5–0.9 µg phage PCR fragments) and an indicated amount of SegB endonuclease. SegB cleavage was carried out at 30°C for 1 h and was stopped by addition of EDTA to a final concentration of 50 mM followed by cooling in ice. DNA products were immediately analyzed by an electrophoresis in 1% agarose gel followed by ethidium bromide staining. One unit (U) of the SegB activity is an amount of the enzyme required for a 50% cleavage of 1 µg DNA of the linearized plasmid pURB1sB at 30°C for 1 h. The specific activity of the SegB preparation used in this work is 1 U/ng.
Identification of the cleavage sites and the recognition site boundaries of SegB endonuclease
To identify cleavage points of SegB endonuclease, 2.5 µg of denatured plasmid pUT2LsB that contains T2L tRNA gene region was annealed with 1 pmol of indicated oligonucleotide. The annealed DNA was labeled in the presence of [α-32P]dATP according to the protocol of ‘Sequenase version 2.0’ DNA sequencing kit (GE Healthcare). To extend annealed primer, 1 µl of 2 mM dNTP was added to 8 µl of the labeled mixture and the incubation was continued for additional 10 min, followed by the inactivation of the Sequenase by heating at 65°C for 20 min. Labeled DNAs produced in 2 µl of the above reactions were cleaved with 2.5 U endonuclease SegB (or with 0.1 U of the enzyme when cleavage points in the major site within T2L tRNAAsn gene were analyzed) in a total volume of 10 µl of the standard SegB cleavage mixture at 30°C for 1 h. Cleavage points of SegB endonuclease located in both strands of T2L tRNAAsn gene site were determined using a labeled DNA in which the upper or lower strand was extended from the indicated oligonucleotide. In all other cases, the position of DNA break in the lower strand was determined indirectly based on the change of the length of SegB cleavage products of the upper strand after their resection with Klenow fragment. To achieve this, a reaction with SegB cleavage of the upper strand was inactivated by heating at 65°C for 20 min, and 5 µl of this reaction mixture supplemented with 1 µl of 0.5 mM dNTP and with 1 U of Klenow fragment (Fermentas, Lithuania) was incubated at 37°C for 30 min. The DNA polymerization reaction was stopped by addition of 3.5 µl Stop solution (95% formamide, 20 mM EDTA, 0.05% bromophenol blue and 0.05% xylenecyanol FF). The SegB cleavage products along with the DNA sequencing ladders generated using the same oligonucleotide were separated in a denaturing 6% PAG.
The following oligonucleotides were used to determine the SegB endonuclease cleavage points in T2L tRNA genes: #6 and #7 for the upper and lower strands of tRNAAsn gene, respectively; #8–13 for tRNAGln, tRNALeu, tRNAGly, tRNAIle, tRNATyr and tRNAArg genes, respectively; and #14 for tRNAPro and tRNASer genes.
SegB endonuclease recognition site located in tRNAAsn gene was determined by the method of Wenzlau et al. (24) using oligonucleotides #6 and #7 for the extension of the upstream and downstream strands, respectively. To define the site, the four sequencing ladder reactions were first produced according to the manufacturer's protocol (Sequenase version 2.0 DNA sequencing kit, USB), except that the termination reactions were stopped by heating at 65°C for 20 min. One microliter of BSA (1 mg/ml), 1 µl of 0.2% Triton X-100 and 2.5 U of SegB endonuclease were then added to each 6 µl termination reaction, and the SegB cleavage was carried out at 30°C for 2 h. The reactions were stopped by addition of 6 µl of the Stop solution (Sequenase version 2.0 DNA sequencing kit, USB) and analyzed along with the control reactions by separation in a denaturing 6% PAG.
Analysis of SegB endonuclease activity on phage DNA
Preparation of phage stocks, purification of phages in a cesium chloride gradient and isolation of phage DNA was carried out as described by Carlson and Miller (25). Phage DNA [(0.5 µg, intact or cleaved by restriction endonuclease SmiI (Sibenzyme)] was treated with 0.2–6 U of the SegB endonuclease. The cleavage products were separated in a 1% agarose gel by field inversion gel electrophoresis (FIGE) in 0.5× TBE buffer using a FIGE Mapper System (Bio-Rad) at forward and reverse voltages of 180 V and 120 V, respectively. Electrophoresis was carried out for 14 h with a linear change of forward and reverse pulse durations from 0.4 s up to 0.1 s. The λDNA digest with HindIII and MassRulerTM DNA Ladder (Fermentas) were used as the size markers. After the electrophoresis, DNA fragments were visualized by staining with ethidium bromide. DNA fragments containing a tRNA gene cluster were identified by a Southern hybridization as described by Sambrook et al. (19). 32P-labeled PCR-amplified DNA fragments produced with pairs of primers #1 and #26, and #1 and #18 were used as probes for phage T2L and phage T4, respectively. The PCR fragments were labeled using a DECAprime II DNA Labelling Kit (Ambion) followed by the removal of the non-incorporated label with a QIAquick Nucleotide Removal Kit (Qiagen).
Crosses of T4-related phages
To cross T4-related phages, E. coli B40 cells were grown in LB media supplemented with 2% casamino acids at 37°C to a density of 5 × 108 cells/ml. An equal volume of mixture containing two phages of interest with a titer of 2.5 × 109 phage particles/ml each was added to a 1 ml of the culture, and the infected culture was incubated without shaking for 5 min at room temperature. The culture was then incubated at 150 r.p.m. for 1 h at 37°C. To complete cell lysis, the culture was added 20 µl chloroform (1/100 part), and the incubation continued for additional 20 min. Cell debris was removed by centrifugation.
To determine inheritance of amber mutation in gene 14, progeny of the mixed infections was plated on the E. coli BE (Sup°) and B40 (SuI) strains. The percentage of progeny containing amber mutation was calculated as the difference of the total progeny (progeny that grows on SuI strain) and the wild-type progeny (progeny that grows on the Sup° strain) over the total progeny multiplied by 100.
To analyze the inheritance of segB gene, Southern analysis of genomic DNAs of progeny of the mixed infections was carried out as follows. The progeny was plated on E. coli B40 (SuI) cells. Individual plaques were then randomly picked and streaked onto the fresh lawn of the E. coli B40 (SuI) cells so that progeny of each plaque was grown on a same sector of three plates. Phage DNA from each of the three plates was transferred onto a nylon HybondN+ membrane (GE Healthcare) and analyzed by hybridization with a 32P-labeled DNA probe complimentary to segB ORF, T2L trna.1 ORF or T2L trna.2 ORF DNA. About 200 progeny plaques were analyzed in each experiment. The percentage of phages containing segB ORF was calculated as a ratio of the number of phage plaques, which gave positive signal in hybridization with the segB ORF-specific probe, to the total number of phage plaques, analyzed in the given hybridization, multiplied by 100.
32P-labeled DNA probes were generated by PCR with oligonucleotides #16 and #21 (T4 segB ORF-specific DNA probe), with oligonucleotides #16 and #21 (T2L trna.1 ORF-specific DNA probe), or with oligonucleotides #24 and #25 (T2L trna.2 ORF-specific DNA probe) in the presence of [α-32P]dATP.
RESULTS
T4-related phages T2L, RB1 and RB3 lack segB gene
Based on the sequence analysis, putative product of segB gene was predicted to be a homing endonuclease of the GIY-YIG family (13). ORFs of homing endonucleases are often absent from otherwise homologous loci of closely related organisms (11,12,15,26). Moreover, those loci frequently contain cleavage site of the homing endonuclease whose ORF is lacking. We first studied whether there are T4-related phages that do not contain segB gene. DNA sequencing analysis of PCR fragments amplified with primers #1 and #26 identified that tRNA gene regions of phages T2L, RB1 and RB3 are highly homologous to that of phage T4 but lack segB gene and its flanking sequences (Figure 1). The sequence analysis also revealed that the phage T2L tRNA gene region contains two ORFs of unknown functions and that DNA sequences of the tRNA gene regions of phages RB1 and RB3 are almost identical (Figure 1).
Figure 1.

Maps of the tRNA gene regions of phages T4, T2L, RB1 and RB3. tRNA genes shared by the four phage phages are shown as the horizontal gray arrows. The horizontal hatched arrows (tRNA genes) and boxes (RNA species I and II genes) represent genes lacking in phage T4 or encoding isoacceptor tRNAs. ORFs are represented by the horizontal open arrows. The vertical arrows show position of the preferred SegB cleavage site in the tRNAAsn gene of phages T2L, RB1 and RB3, and the numbered horizontal arrows show positions of the primers used to produce DNA substrates for characterization of SegB endonuclease activity. The percentages shown above the horizontal gray lines are identities on the DNA sequence level between the indicated T2L or RB1 (RB3) region and the same region of phage T4.
SegB protein is a site-specific endonuclease
Homing endonucleases cleave DNA of homologous loci that lack their cognate ORFs in a site-specific manner (1). In order to analyze whether SegB is a site-specific endonuclease, we placed 666 bp segB ORF under the control of T7 promoter in pET-15b vector. SegB protein was overproduced in E. coli T7lac promoter system, followed by its purification on phospho-cellulose, Ni–NTA, hydroxyapatite and MonoS columns (Figure 2). This purification procedure yields SegB in a near homogeneous form (Figure 2, lane 3). Next, we incubated the purified protein with linearized plasmid pURB1sB DNA that carries phage RB1 tRNA gene cluster lacking SegB ORF and analyzed products of the reactions by gel-electrophoresis in a native agarose gel (Figure 3). The analysis showed that SegB contains a site-specific endonuclease activity that cleaves DNA by introducing double-strand breaks in a reaction requiring Mg2+, Mn2+ or Ca2+ cations. In the presence of Mg2+ or Ca2+ cations, SegB cleaves the RB1 DNA at a single site yielding 2.2 kb and 1.6 kb fragments whereas in the presence of Mn2+ ions (10 mM) SegB cuts the substrate at multiple sites (Figure 3). The multiple cutting was also observed at Mn2+ ions concentration of 1 mM, but to a lesser degree then at 10 mM (data not shown). The SegB endonuclease is not active in the absence of divalent cations or in the presence of Zn2+ cations. An optimal concentration of Mg2+ cations for SegB endonuclease activity is within the range of 9–15 mM (data not shown). Zn2+ cations inhibit the Mg2+-dependent endonuclease activity at a concentration of 50 µM and higher (data not shown).
Figure 2.
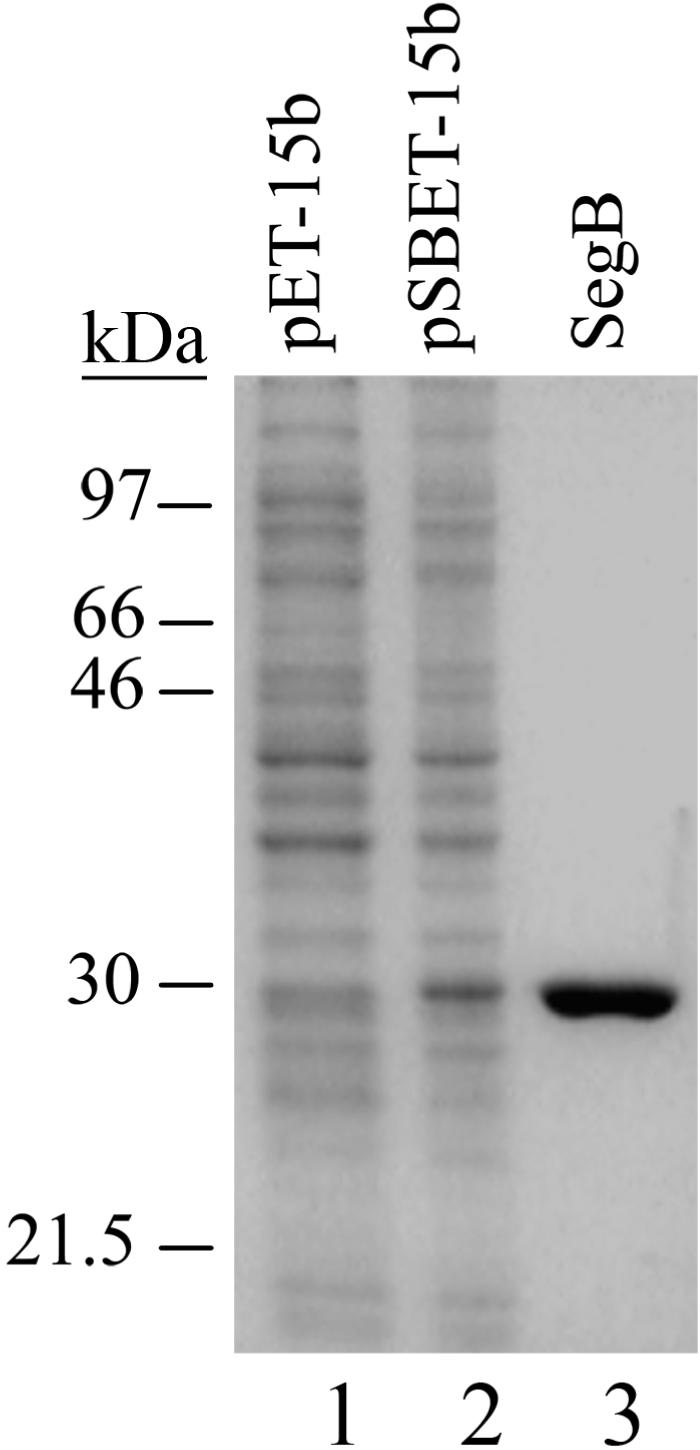
Overproduction and purification of phage T4 SegB protein. Proteins were separated in a 13% PAG followed by Coomassie R-250 staining. The lanes designated as pET-15b and pSBET-15b were loaded with clarified cell-free extracts of BL21(DE3)/pLysE cells, transformed with the corresponding plasmids and grown for 4 h after the induction. SegB lane contains 0.7 µg of the purified protein. Overproduction and purification of T4 SegB protein was carried out as described in ‘Materials and Methods’ section.
Figure 3.
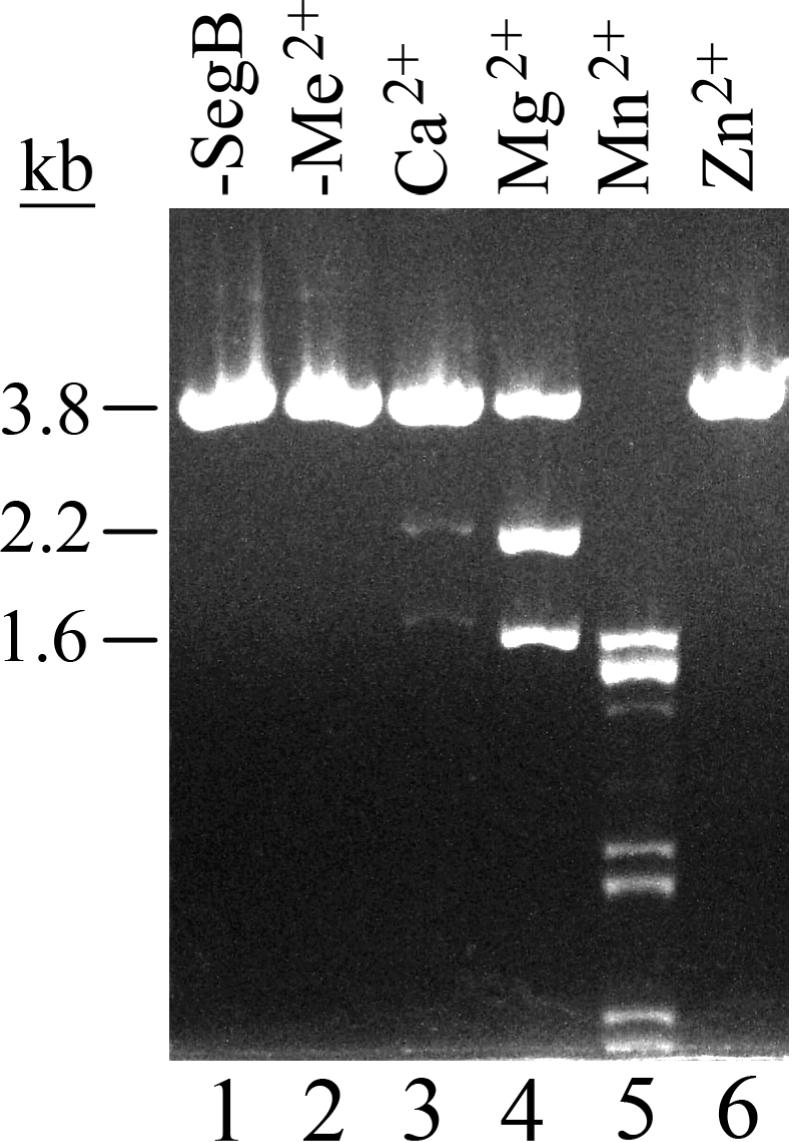
Phage T4 SegB protein possesses an endonuclease activity that cleaves DNA in the presence of Mg2+, Mn2+ or Ca2+ cations. Analysis of SegB endonuclease activity was carried out as described in ‘Materials and Methods’ section, but linearized plasmid pURB1sB, containing the phage RB1 tRNA gene cluster, was used as a substrate and an indicated divalent cation at a final concentration of 10 mM was included in the reaction. Lane ‘-SegB’ displays DNA of a standard reaction from which SegB protein was omitted; Lane ‘-Me2+’ contains DNA products of a standard SegB reaction that lacked divalent cations.
Endonucleases of the GIY-YIG family cleave DNA with relaxed specificity: some sites are highly preferable for endonuclease cleavage, but cleavage also occurs in other sites (15,27). The latter effect is more pronounced when concentration of the enzyme or incubation time is increased. To study whether SegB cuts DNA with relaxed specificity, we cleaved DNA fragments, containing the entire cluster of tRNA genes of phage T2L or phage T4, with 1 U or 2 U of SegB in the presence of physiologically relevant Mg2+ cations (Figure 4). Analysis of the cleavage revealed that T2L tRNA gene region contains a major cleavage site of SegB endonuclease that is efficiently cleaved by 1 U of the enzyme (Figure 4, lane 5). Mapping of the site showed that it is located within T2L tRNAAsn gene (data not shown). In addition to this site, SegB cleaves DNA at many other sites, located ∼70–90 bp from one another, that are less preferable for the cleavage. This is especially well illustrated in the reactions with 2 U of SegB (Figure 4, lanes 8–10). The results of these experiments suggest that SegB cleaves DNA with a relaxed specificity. Because the size of a tRNA gene is about 90 bp, we hypothesized that SegB may cleave DNA at the sites, each of which is located within a tRNA gene, and we confirmed this in experiments presented below.
Figure 4.

SegB endonuclease cleaves DNA at sites that have different susceptibility for the enzyme cleavage. Lanes T2La, T4 and T2Lb contain products of SegB endonuclease cleavage reactions that used a phage T2L fragment, amplified with primers #1 and #2, a phage T4 fragment, amplified with the same primers, and a phage T2L fragment amplified with primers #1 and #5, respectively, as substrates. Locations of the primers #1, #2 and #5 are shown in Figure 1. The cleavage reactions were carried out under the standard conditions, but the amount of SegB endonuclease varied as indicated. Lanes M contain HyperLadder I DNA markers (Bioline). The arrowheads show positions of the 1.24 kb and 0.71 kb, and 0.59 kb and 0.46 kb SegB products cleaved in the sites of T2L tRNAAsn gene (lanes 5 and 8) and of T2L tRNASer gene (lanes 7 and 10), respectively.
Characterization of SegB endonuclease cleavage points and recognition site
To define sequence elements recognized by SegB endonuclease, we first determined positions of SegB cleavage points formed during the hydrolysis of the preferable site located within T2L tRNAAsn gene (Figure 4, lane 5). SegB introduced three cleavage points into either strand of the preferable site (Figure 5A, lane 2). These cleavage points form three double-stranded breaks, each of which has 3′ overhangs of 2 nt long (Figure 5B). To determine SegB recognition sequence, we utilized method of Wenzlau et al. (24) that allows identification of the boundaries of the recognition site. This method is based on endonuclease digestion of four sequencing ladder reactions produced with the primer annealed upstream or downstream of the cleavage site. The sequencing products, which do not contain recognition sequence, are protected from the cleavage by endonuclease. Thereby, for each strand, the 3′-boundary of the recognition site maps after the nucleotide next to the protected region as it defines a minimal sequence required for cleavage. As analysis showed, the recognition site of SegB is 27 bp long, asymmetric with the respect to the cleavage points and is located entirely within T2L tRNAAsn gene (Figure 5A and B).
Figure 5.

Characterization of the major site of SegB endonuclease located within the phage T2L tRNAAsn gene. (A) Images of DNA sequencing gels of the upper and lower strands are presented. Lanes 1 and 2 contain labeled extended DNA products before and after incubation with SegB endonuclease, respectively. Lanes G, A, T and C contain DNA sequencing products obtained in the presence of the corresponding terminator that were treated or not with endonuclease SegB as indicated. (B) DNA sequences surrounding the SegB cleavage site are shown. The cleavage points in the upper and lower strands are shown with closed and open arrowheads, respectively, and the major cleavage points are circled. The minimal sequence required for SegB endonuclease cleavage, which was deduced from the analysis presented in (A), is in capital letters.
As we determined, SegB introduces three double-stranded breaks at the site located within T2L tRNAAsn gene (Figure 5B). We suggest that SegB preferably cuts this substrate at the site distal to the 5′-end of the recognition sequence. As a result, only one of two cleavage products retains the main part of SegB recognition sequence and can be re-cut by SegB at the middle or, less preferably, proximal cleavage sites. This suggestion is based upon analysis of cleavage products ratio when using different amounts of SegB in the reaction mixture. For the top strand, wherein labeled at the 5′-end cleavage products after initial cut at the distal site contain the main part of SegB recognition sequence, number of breaks at the proximal cleavage site relative to two other sites was increased when higher SegB concentration was used (Figure 5A, top strand, lane 2 and lanes ‘+SegB’, 0.1 U and 2.5 U of SegB, correspondingly). And vice versa, lowering of SegB concentration or incubation time resulted in a decrease of cleavage at the proximal site relative to the distal one (data not shown). On the other hand, ratio of cleavage products in the bottom strand was independent from the amount of SegB in the reaction mixture (Figure 5A, bottom strand, lane 2 and lanes ‘+SegB’, 0.1 U and 2.5 U of SegB, correspondingly), where the major break was always at the distal site (respective to the 5′-end of the recognition sequence on the top strand). In this case, labeled cleavage products lost SegB recognition sequence, and, consequently, could not be re-cut by SegB, thereby reflecting real SegB cleavage preference.
We also determined the precise positions of SegB cleavage sites that were detected in our previous experiment and are located in the T2L DNA about 90 nt from one another (Figure 4, lanes 7 and 10). These sites were found to be located within phage T2L tRNAGln, tRNALeu, tRNAGly, tRNAPro, tRNASer, tRNAMet, tRNATyr and tRNAArg genes (Figure 6). With the exception of the upper strand of T2L tRNAGln site, SegB endonuclease introduces more than one cleavage point into the upper or lower strand of each of these sites. This suggests that SegB possesses certain flexibility as to where to introduce cleavage points.
Figure 6.

Identification of additional cleavage sites of endonuclease SegB located in the phage T2L tRNA gene region. Images of the DNA sequencing gels of the upper strand of indicated tRNA genes are presented. The labeled extended DNA was incubated without (lane E) or with endonuclease SegB (lane 1), or with endonuclease SegB followed by DNA extension with Klenow fragment to determine the cleavage points in the lower strand (lane 2). Products of the four DNA sequencing reactions designated as G, A, T and C that were generated with the same primer were used as the ladders. The analyses were carried out as described in ‘Materials and Methods’ section. The sequences surrounding the cleavage sites are shown; positions of the DNA breaks in the upper and lower strands are indicated with closed and open arrowheads, respectively; the major strand break points are circled.
We next aligned the minimal sequence, which SegB recognizes to cleave DNA at the major site within the T2L tRNAAsn gene, with DNA sequences that surround the SegB cleavage sites located in the other T2L tRNA genes (Figure 6) or in the T4 tRNASer gene [the cleavage at the latter site is evident in our previous experiment (Figure 4; lanes 6 and 9) and its precise position was determined using the approach described in Figure 6 (data not shown)]. Alignment of these DNAs allowed us to define the consensus recognition sequence for SegB endonuclease at these ten sites (Figure 7). The consensus sequence contains in its 5′ part GGTTCRANTCC motif, of which 6 nt are 100% conserved. The middle part of the consensus sequence is degenerate, and the 3′ end often contains CCAR sequence immediately followed by preferable SegB cleavage point. In the absence of CCAR sequence, preferable cleavage point tends to be more proximal to the 5′-boundary of the recognition site.
Figure 7.

Identification of conserved nucleotides in the SegB endonuclease recognition site. The minimal recognition sequence of SegB endonuclease (Figure 5) and DNA sequences surrounding the nine SegB endonuclease cleavage sites (Figure 6) were aligned using ClustalW program, version 1.7 (28). Nucleotides that are identical in the all analyzed sequences are highlighted in dark gray; and nucleotides that are the same in no <70% of the sequences are in light gray. These conserved nucleotides were used to define consensus recognition site of SegB endonuclease presented at the bottom. The arrowheads indicate the major SegB cleavage points at the upper strands of the analyzed sites. Nucleotides of the preferable SegB site in the T2L tRNAAsn gene that differ from those of the less preferable sites located in T2L tRNASer and T4 tRNASer genes are boxed.
Of importance is the observation that the DNA recognition sequence of the preferable SegB site within the T2L tRNAAsn gene differs from that of the less preferable site located in the T2L tRNASer gene by 5 nt: G6, A16, G18, A26 and A27 (Figure 7). This suggests that these nucleotides located within the variable part of the consensus recognition sequence are important for making SegB site preferable for the endonuclease cleavage.
SegB endonuclease cleaves the modified DNA of T-even phages
Genomic DNA of T-even phages contains glycosylated 5-hydroxymethyl cytosine instead of cytosine (29). Furthermore, T2 and T4 differ from each other in the character and extent of glycosylation of their DNAs (30). To study whether the cytosine modifications affect SegB endonuclease activity, SegB cleavage of glycosylated 5-hydroxymethyl cytosine DNAs of phages T4 and T2L, 5-hydroxymethyl cytosine DNAs of phage T4290, and cytosine DNA of phage T4alc7 was examined. As shown in Figure 8A, all these genomic DNAs are cleaved with a similar efficiency. To determine whether SegB cleaves glycosylated 5-hydroxymethyl cytosine DNA of phage T2L at the previously identified sites of the tRNA gene region (Figure 4–6), we analyzed genomic T2L DNA cleaved with SegB (Figure 8A) by a Southern analysis with a 32P-labeled probe complimentary to the T2L gene tRNA region (Figure 8B). We found that SegB cleaves the modified DNA of T2L at the tRNAAsn and tRNASer sites with the former one being more preferable for the cleavage. Thus, the glycosylated 5-hydroxymethyl cytosine does not affect the cleavage property of SegB.
Figure 8.

Cleavage of T-even phage DNA by endonuclease SegB is not affected by the presence of cytosine modifications. (A) Glycosylated 5-hydroxymethylcytosine DNA of phages T2L and T4B, 5-hydroxymethylcytosine DNA of T4290, and cytosine DNA of T4alc7 were cleaved under the standard conditions with the indicated amount of SegB endonuclease followed by separation of the DNA products by FIGE in a 1% agarose gel. The SegB endonuclease cleavage and FIGE was carried out as described in ‘Materials and Methods’ section. (B) Glycosylated 5-hydroxymethylcytosine DNA of phage T2L was cleaved under the standard conditions with 1 U of SegB endonuclease. DNA products were separated in a native 1% agarose gel followed by Southern analysis with a 32P-labeled probe complimentary to the phage T2L tRNA gene. Position of the probe is shown in (C). (C) DNA fragment used as 32P-labeled probe in the Southern analysis (B) is represented as gray bar. Positions of SegB cleavage sites located within the tRNASer (Ser) and tRNAAsn (Asn) genes relative to site of SmiI (S) are also shown.
SegB initiates homing of its own ORF in crosses between T4 and T2L
Many T4 genetic markers are inherited preferably by progeny of crosses between closely related phages T4 and T2L (16). Preferential inheritance of at least two of T4 markers is a result of homing events initiated by T4 SegG and SegF homing endonucleases (11,12). Because T4 segB encodes a member of GIY-YIG family of homing endonucleases, we studied whether homing of the segB ORF occurs in crosses between phages T4 and T2L. PCR analysis of 78 randomly picked plaques formed by progeny of the T4 × T2L crosses showed that segB ORF was inherited with 94% efficiency.
In order to analyze whether the endonuclease activity of SegB is required for the preferred inheritance of segB ORF, we introduced a 237 bp deletion within phage B20 segB ORF that removes large part of the presumable catalytic domain of SegB endonuclease. (Phage B20 is a T4 derivative that carries amber mutation in gene 14 located 23 kb from the segB ORF). Using phages B20 and B20▵segB enabled us to study the effects of segB ORF integrity on frequency of its own inheritance. The inheritance of the amber mutation in gene 14 is independent of the segB status and served as the internal control. We found that 99.6% of the progeny of crosses between B20 and T2L contained segB, while the disruption of segB ORF decreased the preferred inheritance of the gene to the level (65%) observed for SegB-independent inheritance of the phage T4 gene 14 (Table 1). These results suggest that the endonuclease activity of SegB promotes homing of its own ORF in crosses between phages T4 and T2L.
Table 1.
Inheritance of segB ORF and gene 14 by progeny of crosses between phages T4amB20 and T2L
| Cross | aFraction of progeny containing segB (%) | bFraction of progeny containing trna.1 ORF (%) | cFraction of progeny containing trna.2 ORF (%) | dFraction of progeny with gene 14 amber mutation (%) |
|---|---|---|---|---|
| B20 × T2L | 99.6 | 0.4 | 0.4 | 65.1 ± 1.6 |
| B20▵segB × T2L | 63 | ND | ND | 65.5 ± 0.6 |
The ratio of parent phages for each cross was 1:1. The 467 progeny plaques of either cross obtained from two independent experiments (about 230 plaques from each experiment) were analyzed by Southern hybridization with a 32P-labeled probe complimentary to aphage T4 segB ORF, bphage T2L trna.1 ORF, cphage T2L trna.2 ORF. dTo estimate progeny fraction carrying gene 14 amber mutation, the progeny obtained from the two independent experiments (about 300 plaques from either experiment) were analyzed for the presence of amber-mutation and the data are shown as averages ± SD. ND, not determined.
Homing of segB ORF into the T2L gene tRNA region must replace part of the T2L region located between tRNASer and rnaI genes (Figure 1), which is nonhomologous to the T4 region. Accordingly, of 467 progeny plaques generated in two independent B20 × T2L crosses, two progeny plaques only hybridized with a 32P-labeled probe complimentary to T2L trna.1 ORF or T2L trna.2 ORF but not with a 32P-labeled probe complimentary to T4 segB ORF (Table 1). These data suggest that segB ORF homing occurring in the crosses of T4 and T2L is accompanied with exclusion of the T2L tRNA gene cluster.
DISCUSSION
Amino acid sequence of bacteriophage T4 SegB protein was found to contain regions that are homologous to homing endonucleases of the GIY-YIG family (13). Yet, biological function of SegB protein has been unknown. segB ORF is located within the cluster of tRNA genes, a genetic region that is not essential for phage T4 development. In the current work, we have found that SegB is a homing endonuclease. SegB is therefore another member of the growing list of homing endonucleases encoded by bacteriophage T4 (14). Why does rather small genome of phage T4 (∼169 kb) carry 14 genes of homing endonucleases? Answer to this question remains unknown, but several hypotheses have been discussed (11,12). One obvious biological role of the homing endonucleases is that these enzymes stabilize the phage T4 genetic information by ensuring that the host genetic information is not lost by progeny of the mixed infections.
By using electron microscopy, Kim and Davidson (31) have found that phages T4 and T2L contain an extended nonhomologous region located in the tRNA gene cluster. As shown in the present work, this nonhomology is due to the fact that phage T2L lacks segB gene and the flanking tRNA genes but instead contains two ORFs of unknown function and four extra tRNA genes (Figure 1). segB ORF is also absent from the genomes of T4-related phages RB1, RB3, RB6, RB7, RB8 and RB9 (Figure 1 and data not shown). These observations are consistent with the idea that T4 genome acquired segB ORF quite recently.
SegB endonuclease cleaves DNA by introducing double-stranded breaks in a reaction that requires the presence of Mg2+, Mn2+ or Ca2+ cations (Figure 3). Because among these three cations, Mg2+ is physiologically relevant cation, we focused on characterization of the Mg2+-dependent endonuclease activity. Like many homing endonucleases including I-TevI and the other members of GIY-YIG family (1), the enzyme cleaves DNA with a relaxed specificity: some sites are highly preferable for the cleavage, but if SegB concentration is increased, the cleavage occurs at other sites (Figure 4).
In the T2L tRNA gene cluster lacking segB ORF we have identified nine SegB cleavage sites, each of which is localized at the 3′ end of a tRNA gene (Figures 5 and 6). Of these sites, the one located within the tRNAAsn gene is the most preferable for SegB cleavage (Figures 4 and 8B). By using the approach of Wenzlau et al. (24), we have determined that SegB endonuclease recognizes a 27 bp sequence to cleave DNA at the tRNAAsn site. The length of the SegB recognition site is comparable to those of the other homing endonuclease recognition sites that are in the range from 12 bp to 40 bp (1–3). Alignment of the SegB recognition sequence with DNA sequences surrounding the other nine SegB cleavage sites strongly suggests that several nucleotide residues within the recognition site are conserved (Figure 7). Notably, GGTTCRANTCC sequence, which appears to be an essential element of SegB recognition site, corresponds to RGUUCRANYCY sequence motif of TψC stem-loop of tRNAs conserved in all biological kingdoms (32). On one side, wide distribution of potential SegB cleavage sites increases probability of segB gene to colonize new species, on the other side, in combination with relaxed specificity; this makes SegB potentially toxic to a host. Bacteriophage T4 contains at least five SegB cleavage sites located within the genes for tRNAGln, tRNALeu, tRNAGly, tRNAPro and tRNASer that is evident from the sequence identity of cognate T4 and T2L genes (Figure 1). Their cleavage in vitro, though to a different extent, depends on the SegB concentration in the reaction mixture (Figure 4). Moreover, naturally modified T4 DNA is not protected from the cleavage by SegB, and can be cleaved in numerous sites when the enzyme is used in excess (Figure 8A). How does T4 protect itself against SegB? One way it could occur is that phage T4 tightly controls the level of expression of segB gene. According to the findings obtained by Broida and Abelson (33) that are in agreement with the data of microarray analysis of phage T4 gene expression (34), transcription of segB gene is primarily directed by the middle promoter, which is located just upstream of tRNA gene cluster. As presumed, the transcription passes through the entire tRNA gene cluster and terminates after species I RNA gene. Therefore, segB mRNA is synthesized as a part of ∼1.75 kb primary transcript, which is also a precursor of eight tRNAs and species I and II RNAs. Analysis of untranslated region at 5′-end of segB ORF together with existing data about the primary transcript processing allowed us to presume two possible ways of negative control of segB expression. The first putative mechanism is occlusion of translation initiation region for segB ORF by stable hairpin (ΔG = 20.2 kcal as calculated by Mfold program; 35) that is capable to be formed at 5′-end of the segB ORF (71912–71952 position of T4 genome). Both Shine-Delgarno sequence and initiation codon of segB ORF are predicted to be in the hairpin stem. The inhibitory effect of hairpin on translation was shown for a number of phage T4 genes (36). This mechanism serves to prevent synthesis of the late T4 proteins from the early transcripts. It is of particular interest to note that such mechanism of translational control was shown for T4 intron-encoded homing endonucleases I-TevI, I-TevII and I-TevIII (37), as well as suggested for T4-like phage Aeh1 homing endonuclease MobE encoded by freestanding ORF (38). Late or middle transcripts for those proteins are initiated from promoters located just upstream or overlapping with the sequestering hairpin. These mRNAs lack a hairpin forming sequence at the 5′-end and, therefore, are active in translation. However, there are no promoters, which map to the close proximity of the 5′-end of segB ORF (14), suggesting that segB mRNA translation remains to be restrained. The second possible mechanism to control segB expression is to impair segB mRNA efficacy by site-specific cleavage between the last and stop codons of segB ORF during primary transcript processing under maturation of tRNAArg (39). It may, on one side, facilitate mRNA degradation, and, on the other side, result in a synthesis of premature inactive protein. Similarly, lowering of gene expression level by primary transcript processing was proposed for phage Aeh1 homing endonuclease MobE, whose transcript was shown to be internally cleaved at an RNase E-like site (38), and for phage T5 homing endonucleases F-TflI, F-TflII and F-TflII, which ORFs are also located in tRNA gene cluster (18 and Ksenzenko,V.N., personal communication). Consistent with the assumption about tight control of the expression, we could not detect the SegB endonuclease activity in cell-free extracts of T4-infected E. coli cells, while activity of SegD, exhibiting higher cleavage specificity, was easily detected (data not shown). Apparently, SegB is synthesized in small amounts that under co-infection of T4 and T2L are sufficient to cleave only T2L DNA at preferable site located in tRNAAsn gene and to initiate genetic exchange of this region.
SegB prefers to introduce major cleavage point 3′ from CCAR sequence (Figure 7). When the CCAR sequence is absent, the cutting occurs closer to the 5′ boundary of the recognition site. This property of SegB is especially well documented in the case of the enzyme sites located within the T2L and T4 tRNASer genes (Figure 7). With the exception of CCAG sequence, the sequences of these two sites are identical. SegB cuts DNA of the T2L tRNASer site immediately downstream from the CCAG, while the cleavage of the T4 tRNASer site lacking the CCAG occurs three nucleotides upstream relative to the T2L site cleavage point. Other endonucleases of GIY-YIG family, I-TevI and I-BmoI, also have a preference to introduce the cleavage points within certain sequences (40–42). Furthermore, for I-TevI it has been shown that small insertions or deletions introduced between the main element of the recognition site and the cleavage site do not change the sequence-specific position of the cleavage (40).
SegB endonuclease cleavage tolerates the glycosylated 5-hydroxyl-cytosine modification that is located in the large groove of the double helix. This modification of DNA also does not affect the cleavage catalyzed by intron-encoded endonucleases I-TevI and I-TevII (8,27). This is consistent with the idea that phage T4 encoded homing endonucleases predominantly contact DNA along the small grooves.
The best studied member of the GIY-YIG family is I-TevI endonuclease encoded by group I intron of T4 thymidylate synthase gene. I-TevI consists of two structural domains, connected by a flexible linker. The N-terminal domain performs the catalytic function, and the C-terminal domain is responsible for DNA recognition and binding (43,44). The DNA-binding domain of I-TevI has an extended structure and consists of three subdomains: a Zn finger, an extended segment with the α-helical site responsible for DNA minor-groove binding and a helix–turn–helix (45). Though it is the recognition domain that is mainly responsible for the DNA target recognition and binding, the catalytic domain of I-TevI still shows some preference for a certain sequence in the cleavage site (40,41,46). As was discussed above, the majority of the SegB enzymatic properties are very similar to I-TevI. Moreover, SegB and I-TevI contain GIY-YIG motif in their N-termini and both proteins contain ‘NUMOD 3’ motif, implicated in DNA binding, in their C-terminal parts (47). Given these similarities in both enzymatic properties and amino acid sequences, we suggest that the domain organization of SegB may be similar to that of I-TevI.
Exclusion of phage T2L genetic markers that occurs in the crosses with phage T4 is mostly characteristic for the genetic markers that are located within regions heterologous to those of phage T4 (16). Molecular mechanism of this phenomenon remained unknown for a long time. A recent work by Belle et al. (11) suggested that homing events occurring in the T4 × T2L crosses is the basis of the exclusion of T2L markers. They showed that the exclusion of T2L gene 56 is the result of gene conversion events initiated by SegF homing endonuclease. Exclusion of another T2L marker, gene 32, is directed by T4 homing endonuclease SegG (12). Similarly, SegE, whose ORF is within a nonessential region, initiates homing of its own gene that results in exclusion of the homologous RB30 region (15).
In their pioneering work, Russell and Huskey (16) were not able to study exclusion of the T2L tRNA gene cluster. This happened because of the unavailability of T4 or T2L genetically tractable markers located in this region, which is not essential for the development of the phages (48). As we have shown in this work, during co-infection of phages T4 and T2L, endonuclease SegB is responsible for an efficient homing of its own ORF into the T2L tRNA gene cluster. This process is apparently initiated by SegB cleavage of the preferable site within T2L tRNAAsn gene and is accompanied with an almost complete exclusion of the T2L DNA region of ∼1250 bp long, which encodes tRNAMet, tRNATyr, tRNAAsn and tRNAArg genes, and also trna.1 and trna.2 ORFs. These results support the idea that the exclusion of most T2L genetic markers observed in the mixed infections is a net result of the action of T4 homing endonucleases (11,49).
ACKNOWLEDGEMENT
We thank Dr. V. Tanyashin for bacterial and phage strains, and for comprehensive help.
Conflict of interest statement. None declared.
REFERENCES
- 1.Belfort M, Roberts R. Homing endonucleases: keeping the house in order. Nucleic Acids Res. 1997;25:3379–3388. doi: 10.1093/nar/25.17.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757–3774. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoddard BL. Homing endonuclease structure and function. Q. Rev. Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 4.Lambowitz AM, Belfort M. Introns as mobile genetic elements. Ann. Rev. Biochem. 1993;62:587–622. doi: 10.1146/annurev.bi.62.070193.003103. [DOI] [PubMed] [Google Scholar]
- 5.Belfort M, Perlman PS. Mechanisms of intron mobility. J. Biol. Chem. 1995;270:30237–30240. doi: 10.1074/jbc.270.51.30237. [DOI] [PubMed] [Google Scholar]
- 6.Edgell DR, Belfort M, Shub DA. Barriers to intron promiscuity in bacteria. J. Bacteriol. 2000;182:5281–5289. doi: 10.1128/jb.182.19.5281-5289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell-Pedersen D, Quirk SM, Bryk M, Belfort M. I-TevI, the endonuclease encoded by the mobile td intron, recognizes binding and cleavage domains on its DNA target. Proc. Natl Acad. Sci. USA. 1991;88:7719–7723. doi: 10.1073/pnas.88.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loizos N, Silva GH, Belfort M. Intron-encoded endonuclease I-TevII binds across the minor groove and induces two distinct conformational changes in its DNA substrate. J. Mol. Biol. 1996;255:412–424. doi: 10.1006/jmbi.1996.0034. [DOI] [PubMed] [Google Scholar]
- 9.Sharma M, Hinton DM. Purification and characterization of the SegA protein of bacteriophage T4, an endonuclease related to proteins encoded by group I introns. J. Bacteriol. 1994;176:6439–6448. doi: 10.1128/jb.176.21.6439-6448.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadyrov FA, Kriukov VM, Shliapnikov MG, Baev AA. SegE – a new site-specific endodeoxyribonuclease from bacteriophage T4. Dokl. Akad. Nauk. 1994;339:404–406. [PubMed] [Google Scholar]
- 11.Belle A, Landthaler M, Shub DA. Intronless homing: site-specific endonuclease SegF of bacteriophage T4 mediates localized marker exclusion analogous to homing endonucleases of group I introns. Genes Dev. 2002;16:351–362. doi: 10.1101/gad.960302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Belle A, Shub DA, Belfort M, Edgell DR. SegG endonuclease promotes marker exclusion and mediates co-conversion from a distant cleavage site. J. Mol. Biol. 2003;334:13–23. doi: 10.1016/j.jmb.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 13.Sharma M, Ellis RL, Hinton DM. Identification of a family of bacteriophage T4 genes encoding proteins similar to those present in group I introns of fungi and phages. Proc. Natl Acad. Sci. USA. 1992;89:6658–6662. doi: 10.1073/pnas.89.14.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadyrov FA, Shlyapnikov MG, Kryukov VM. A phage T4 site-specific endonuclease, SegE, is responsible for a non-reciprocal genetic exchange between T-even-related phages. FEBS Lett. 1997;415:75–80. doi: 10.1016/s0014-5793(97)01098-3. [DOI] [PubMed] [Google Scholar]
- 16.Russell RL, Huskey RJ. Partial exclusion between T-even bacteriophages: an incipient genetic isolation mechanism. Genetics. 1974;78:989–1014. doi: 10.1093/genetics/78.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukada K, Abelson J. DNA sequence of a T4 transfer RNA gene cluster. J. Mol. Biol. 1980;139:377–391. doi: 10.1016/0022-2836(80)90136-9. [DOI] [PubMed] [Google Scholar]
- 18.Akulenko NV, Ivashina TV, Shaloiko LA, Shliapnikov MG, Ksenzenko VN. Novel site-specific endonucleases F-TflI, F-TflII and F-TflIV encoded by the bacteriophage T5. Mol. Biol. 2004;38:632–641. [PubMed] [Google Scholar]
- 19.Sambrook J, Fritch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 20.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 21.Wood WB. Host specificity of DNA produced by Escherichia coli: bacterial mutations affecting the restriction and modification of DNA. J. Mol. Biol. 1966;16:118–133. doi: 10.1016/s0022-2836(66)80267-x. [DOI] [PubMed] [Google Scholar]
- 22.Studier W, Rosenberg A, Dunn J, Dubendorff J. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 24.Wenzlau JM, Saldanha RJ, Butow RA, Perlman PS. A latent intron-encoded maturase is also an endonuclease needed for intron mobility. Cell. 1989;56:421–430. doi: 10.1016/0092-8674(89)90245-6. [DOI] [PubMed] [Google Scholar]
- 25.Carlson K, Miller ES. Experiments in T4 genetics. In: Karam JD, editor. Molecular Biology of Bacteriophage T4. Washington DC: ASM Press; 1994. pp. 427–434. [Google Scholar]
- 26.Sandegren L, Sjoberg BM. Distribution, sequence homology, and homing of group I introns among T-even-like bacteriophages: evidence for recent transfer of old introns. J. Biol. Chem. 2004;279:22218–22227. doi: 10.1074/jbc.M400929200. [DOI] [PubMed] [Google Scholar]
- 27.Bryk M, Quirk SM, Mueller JE, Loizos N, Lawrence C, Belfort M. The td intron endonuclease I-TevI makes extensive sequence-tolerant contacts across the minor groove of its DNA target. EMBO J. 1993;12:2141–2149. doi: 10.1002/j.1460-2075.1993.tb05862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Revel HR. DNA modification: glucosylation. In: Mathews CK, Kutter EM, Mosig G, Begret PB, editors. Bacteriophage T4. Washington DC: American Society for Microbiology; 1983. pp. 156–165. [Google Scholar]
- 30.Lehman IR, Pratt EA. On the structure of the glucosylated hydroxymethylcytosine nucleotides of coliphages T2, T4, and T6. J. Biol. Chem. 1960;235:3254–3259. [PubMed] [Google Scholar]
- 31.Kim JS, Davidson N. Electron microscope heteroduplex study of sequence relations of T2, T4, and T6 bacteriophage DNAs. Virology. 1974;57:93–111. doi: 10.1016/0042-6822(74)90111-1. [DOI] [PubMed] [Google Scholar]
- 32.Dirheimer G, Keith G, Dumas P, Westhof E. Primary, secondary, and tertiary structures of tRNAs. In: Söll D, RajBhandary UL, editors. tRNA: Structure, Biosynthesis, and Function. Washington DC: American Society for Microbiology; 1995. pp. 93–126. [Google Scholar]
- 33.Broida J, Abelson J. Sequence organization and control of transcription in the bacteriophage T4 tRNA region. J. Mol. Biol. 1985;185:545–563. doi: 10.1016/0022-2836(85)90071-3. [DOI] [PubMed] [Google Scholar]
- 34.Luke K, Radek A, Liu X, Campbell J, Uzan M, Haselkorn R, Kogan Y. Microarray analysis of gene expression during bacteriophage T4 infection. Virology. 2002;299:182–191. doi: 10.1006/viro.2002.1409. [DOI] [PubMed] [Google Scholar]
- 35.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller ES, Karam JD, Spicer E. Control of translation initiation: mRNA structure and protein repressors. In: Karam J, Drake JW, Kreuzer KN, Mosig G, Hall DH, Eiserling FA, Black LW, Spicer EK, Kutter E, Carlson K, Miller ES, editors. Molecular Biology of Bacteriophage T4. Washington DC: American Society for Microbiology; 1994. pp. 193–205. [Google Scholar]
- 37.Gott JM, Zeeh A, Bell-Pedersen D, Ehrenman K, Belfort M, Shub DA. Genes within genes: independent expression of phage T4 intron open reading frames and the genes in which they reside. Genes Dev. 1988;2:1791–1799. doi: 10.1101/gad.2.12b.1791. [DOI] [PubMed] [Google Scholar]
- 38.Gibb EA, Edgell DR. Multiple controls regulate the expression of mobE, an HNH homing endonuclease gene embedded within a ribonucleotide reductase gene of phage Aeh1. J. Bacteriol. 2007;189:4648–4661. doi: 10.1128/JB.00321-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt FJ, Apirion D. T4 transfer RNAs: paradigmatic system for the study of RNA processing. In: Mathews CK, Kutter EM, Moesig G, Berget PB, editors. Bacteriophage T4. Washington DC: American Society for Microbiology; 1983. pp. 208–217. [Google Scholar]
- 40.Bryk M, Belisle M, Mueller JE, Belfort M. Selection of a remote cleavage site by I-TevI, the td intron-encoded endonuclease. J. Mol. Biol. 1995;247:197–210. doi: 10.1006/jmbi.1994.0133. [DOI] [PubMed] [Google Scholar]
- 41.Edgell DR, Stanger MJ, Belfort M. Coincidence of cleavage sites of intron endonuclease I-TevI and critical sequences of the host thymidylate synthase gene. J. Mol. Biol. 2004;343:1231–1241. doi: 10.1016/j.jmb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Edgell DR, Stanger MJ, Belfort M. Importance of a single base pair for discrimination between intron-containing and intronless alleles by endonuclease I-BmoI. Curr. Biol. 2003;13:973–978. doi: 10.1016/s0960-9822(03)00340-3. [DOI] [PubMed] [Google Scholar]
- 43.Derbyshire V, Kowalski JC, Dansereau JT, Hauer CR, Belfort M. Two-domain structure of the td intron-encoded endonuclease I-TevI correlates with the two-domain configuration of the homing site. J. Mol. Biol. 1997;265:494–506. doi: 10.1006/jmbi.1996.0754. [DOI] [PubMed] [Google Scholar]
- 44.Dean AB, Stanger MJ, Dansereau JT, Van Roey P, Derbyshire V, Belfort M. Zinc finger as distance determinant in the flexible linker of intron endonuclease I-TevI. Proc. Natl Acad. Sci. USA. 2002;99:8561. doi: 10.1073/pnas.082253699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Roey P, Waddling CA, Fox KM, Belfort M, Derbyshire V. Intertwined structure of the DNA-binding domain of intron endonuclease I-TevI with its substrate. EMBO J. 2001;20:3631–3637. doi: 10.1093/emboj/20.14.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller JE, Smith D, Bryk M, Belfort M. Intron-encoded endonuclease I-TevI binds as a monomer to effect sequential cleavage via conformational changes in the td homing site. EMBO J. 1995;14:5724–5735. doi: 10.1002/j.1460-2075.1995.tb00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sitbon E, Pietrokovski S. New types of conserved sequence domains in DNA-binding regions of homing endonucleases. Trends Biochem. Sci. 2003;28:473–477. doi: 10.1016/S0968-0004(03)00170-1. [DOI] [PubMed] [Google Scholar]
- 48.Wilson JH, Kim JS, Abelson JN. Bacteriophage T4 transfer RNA. 3. Clustering of the genes for the T4 transfer RNAs. J. Mol. Biol. 1972;71:547–556. doi: 10.1016/s0022-2836(72)80022-6. [DOI] [PubMed] [Google Scholar]
- 49.Goodrich-Blair H, Shub DA. Beyond homing: competition between intron endonucleases confers a selective advantage on flanking genetic markers. Cell. 1996;84:211–221. doi: 10.1016/s0092-8674(00)80976-9. [DOI] [PubMed] [Google Scholar]