

SUMMARY
Prodrug gene therapy (PGT) is a treatment strategy in which tumor cells are transfected with a 'suicide' gene that encodes a metabolic enzyme capable of converting a nontoxic prodrug into a potent cytotoxin. One of the most promising PGT enzymes is cytosine deaminase (CD), a microbial salvage enzyme that converts cytosine to uracil. CD also converts 5-fluorocytosine (5FC) to 5-fluorouracil (5FU), an inhibitor of DNA synthesis and RNA function. Over 150 studies of cytosine deaminase-mediated PGT applications have been reported since 2000, all using wild-type enzymes. However, various forms of cytosine deaminase are limited by inefficient turnover of 5FC and/or limited thermostability.
In a previous study we stabilized and extended the half-life of yeast cytosine deaminase (yCD) by repacking of its hydrophobic core at several positions distant from the active site. Here we report that random mutagenesis of residues selected based on alignment with similar enzymes, followed by selection for enhanced sensitization to 5FC, also produces an enzyme variant (yCD-D92E) with elevated Tm values and increased activity half-life. The new mutation is located at the enzyme's dimer interface, indicating that independent mutational pathways can lead to an increase in the temperature that induces protein unfolding and aggregation in thermal denaturation experiments measured by circular dichroism spectroscopy, and an increase in the half-life of enzyme activity at physiological temperature, as well as more subtle effect on enzyme kinetics. Each independently derived set of mutations significantly improves the enzyme's performance in PGT assays both in cell culture and in animal models.
Keywords: thermostability, prodrug gene therapy, cytosine deaminase, random mutagenesis, structure
INTRODUCTION
The pyrimidine salvage pathway enzyme cytosine deaminase (CD; EC 3.5.4.1) is responsible for converting the nucleobase cytosine to uracil and ammonia. This activity is found primarily in microbes1, including Saccharomyces cerevisiae and Escherichia coli, and has arisen at least twice, using completely separate protein folds2,3. In addition to cytosine, cytosine deaminase catalyzes the conversion of 5-fluorocytosine (5FC) to the potent chemotherapeutic drug, 5-fluorouracil (5FU). Thus, the combination of CD enzyme activity and 5FC as its substrate forms the basis of a potential anti-tumor gene therapy, where CD plays the role of a 'suicide gene'4.
In suicide gene therapy applications, the gene for cytosine deaminase is introduced into cancer cells, followed by systemic administrations of the prodrug 5FC. Following deamination by CD, 5FU is converted by cellular enzymes to 5FdUMP, an irreversible inhibitor of thymidylate synthase (TS). Inhibition of TS blocks dTTP production and prevents DNA synthesis5–7. The CD/5FC system has been used in numerous animal models8–10 and is currently being evaluated in clinical trials for solid tumors11–15. One limitation to this approach is the poor transfection efficiency of current vector delivery systems. Consequently, high 5FC doses must be administered to achieve therapeutic value and are associated with unwanted side effects suggested to be a result of the generation of 5FU by intestinal bacteria16.
Key to suicide gene therapy is the phenomenon known as the bystander effect, in which non-transfected neighboring cells are killed through the transfer of antimetabolites from CD expressing cells in close proximity. A strong bystander effect has been associated with CD and 5FC because 5FU is a small, uncharged molecule capable of non-facilitated diffusion through cellular membranes17–21. Unlike other suicide gene therapy systems such as herpes simplex virus thymidine kinase (HSVTK) and ganciclovir (GCV) that rely on transfer of metabolites through gap junctions, the CD/5FC bystander effect is not dependent upon cell-to-cell contact17.
Another advantage of the CD/5FC combination is that 5FU has radiosensitizing properties22–23. Since it is unlikely that treatment with gene therapy would be the only course of action in patients, radiosensitizing effects can augment treatment regimens. Several groups have reported in vivo results with CD to have a significant bystander effect22.24 at clinically relevant 5FC doses and radiation regimens22,25–26.
Two completely separate forms of CD have evolved in nature, and both are being studied in anti-tumor gene therapy investigations. Yeast CD (yCD) belongs to the amidohydrolase protein fold family (CATH topology class 3.40.140) and shares homology with bacterial and eukaryotic cytidine deaminases3. The enzyme is assembled into a homodimer comprised of 17.5 kDa subunits that contain a catalytic zinc ion. In contrast, bacterial CD (bCD) belongs to the alpha-beta ‘TIM’ barrel fold family (CATH topology class 2.30.40) and most closely resembles human adenosine deaminase2. In E. coli, the enzyme is assembled into a hexamer of 60 kDa subunits that contain a catalytic iron.
Although product release from the yeast cytosine deaminase is rate-limiting27, yCD has been observed to display superior kinetic properties towards 5FC (corresponding to a 22-fold lower Km for the prodrug) and slightly improved efficacy for treating tumors in mice than bCD28. However, wild-type yCD is relatively thermolabile as compared to bCD28–29, a property that may limit its performance in therapeutic applications.
Using computational protein engineering, we previously created a series of mutant yCD variants that display elevated unfolding temperatures in denaturation experiments and increased half-lives of catalytic activity at elevated temperatures30. An enzyme mutant with two substitutions (A23L/I1140L or 'yCD-double') and a subsequent, final redesigned mutant (A23L/V108I/I140L or 'yCD-triple') display wild-type catalytic efficiencies, and 5- and 30-fold increases in the half-lives of those catalytic activities. Furthermore, yCD-double and yCD-triple display apparent melting temperatures 6°C and 10°C greater, respectively, than wild-type yCD. The latter construct also displayed improved complementation of cytosine deaminase activity at elevated temperatures in bacteria.
In the present study, we have randomly mutagenized residues that were selected based on sequence conservation with similar enzymes, and have used positive and negative genetic complementation strategies to identity mutations that confer increased 5FC sensitivity (i.e., that lead to toxicity at the lowest possible concentrations of 5FC). The resulting mutations were analyzed alone and in combination with those previously engineered in the protein core for their effect on (1) prodrug sensitivity in mammalian cells and in a mouse xenograft tumor model, (2) substrate specificity, (3) kinetic efficiency, and (4) thermal unfolding of the enzyme and the half-life of catalytic activity. The crystal structure of the mutant from the screen that induced the most significant increase in 5FC sensitivity was determined. The computationally engineered yCD-triple and the genetically selected yCD-D92E variant each separately provide enhanced 5FC-sensitivity to cells and therefore may serve as improved candidates for future suicide gene therapy studies. In contrast, the combination of these mutations abrogates their individual contribution to improved performance in PGT assays, possibly due to the accumulation of individual small reductions in catalytic efficiencies that reach a critical threshold for sensitization to 5- FC.
RESULTS
Identification of mutants in and near the active site that confer enhanced 5FC sensitivity
To create yCD variants with increased activity to 5FC, we subjected 11 codons within the most conserved region of yCD (T83, L84, Y85, T86, L88, S89, D92, M93, T95, G96 and I98) to regio-specific, partially randomizing mutagenesis as described in Materials and Methods. Absolutely conserved residues within this same region, assumed to be critical for activity, were omitted from randomization (T87, P90, C91 and C94). Figure 1 shows a model of two different views of yCD (purple residues indicate the locations of the triple amino acid substitutions, A23L/V108I/I140L) with targeted residues highlighted in blue.
Figure 1.
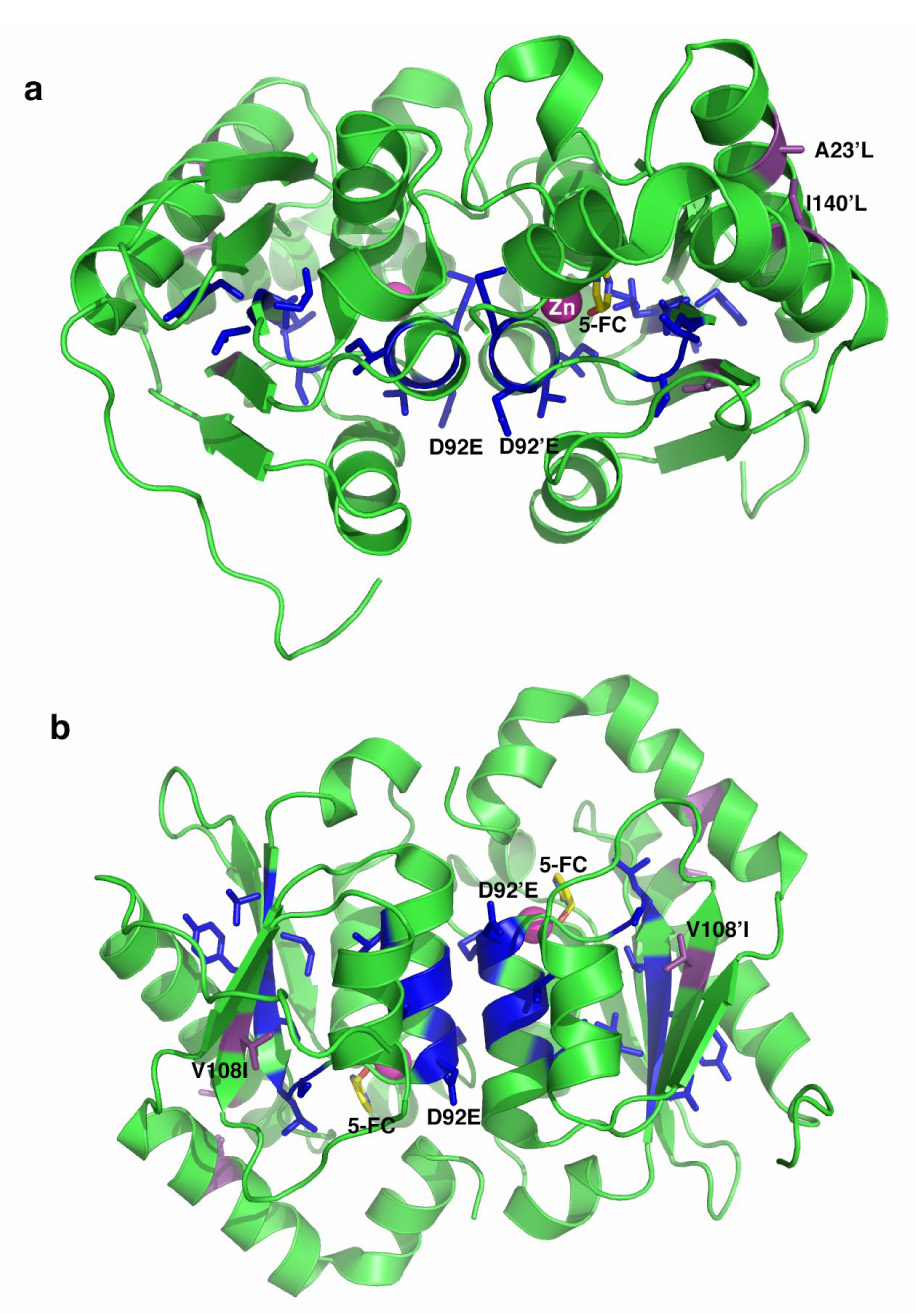
Ribbon diagram of the yCD homodimer (a) side view, b) bottom view) showing the location of eleven residues that were subjected to randomization and subsequent selection for 5FC sensitization (side chains shown as blue sticks) and three residues that were previously redesigned to increase packing between structural elements and thus increase thermostability (shown in purple). The location and nature of mutations characterized in this paper (D92E from selection; A23L, V108I, and I140L) are labeled. Each mutation is found once in each protein subunit.
Identification of functional yCD variants with enhanced 5FC activity involved a two-step selection procedure. First, functional yCD variants were selected for preservation of catalytic activity, based upon their ability to confer growth on cytosine-containing plates, under conditions that require CD activity for viability. Second, the functional variants were then screened on cytosine plates containing 5FC. It was determined that the 5FC dose at which wild-type yCD survives, or the sub-lethal dose, is 77.45µM. Although wild-type pETHT:yCD will allow growth on plates containing the sub-lethal dose of 5FC, any mutant with increased 5FC activity will not be viable. To identify the best variants within the library pool colonies were sequentially streaked onto plates containing 5FC from 77.45µM to 3.87µM. From an estimated total of 34,000 transformants, 50 colonies (~0.15%) were identified as yCD positive based on their ability to grow on cytosine containing plates. Of the 50 yCD positive clones, only six conferred sensitivity at 3.87µM 5FC, the lowest effective 5FC concentration in the negative selection.
In order to evaluate the library diversity, along with identifying which substitutions are tolerated, plasmid DNA from colonies on the non-selective and selective plates was isolated and sequenced. Sequence analysis of DNA isolated from colonies that grew on the non-selected plates revealed a broad spectrum of mutations in the regio-specific library (data not shown) and indicates that the oligonucleotides used to generate the library contained random sequences. Sequence analysis of the 6 variants that conferred the greatest sensitivity to E. coli revealed that two had a substitution at D92 to glutamic acid (D92E), two had a substitution at M93 to leucine (M93L) and two had a substitution of I98 to leucine (I98L). Additionally, the two I98L mutants have different nucleotide-level changes, suggesting these mutants were derived independently. Therefore, from a library of 34,000 transformants, three amino acid substitutions were identified that confer 5FC sensitivity (D92E, M93L, and I98L) at the lowest 5FC concentration in the selection scheme.
Construction of combinations of yCD mutants
Recently, we performed a computational design study aimed at improving the stability of the thermolabile yCD30, in which mutations were generated in the enzyme's hydrophobic core, distant from the active site. To investigate the combined effect of these mutations with those selected from the mutagenesis study as described above, several new mutants were constructed by site-directed mutagenesis. These 'superimposed' mutants (yCD-triple/D92E, yCD-triple/M93L and yCD-triple/I98L), and their individual parental mutations, were then tested in a mammalian tumor cell line assay for increased 5FC sensitivity.
In vitro 5FC sensitivity assays
To determine the activity of these mutants towards 5FC (and their ability to induce sensitivity to 5FC) in vitro, mammalian expression vectors encoding the yCD variants were constructed and used to transfect rat C6 glioma cells (see Materials and Methods). Immunoblot analyses of lysates from the pools of transfectants show similar yCD expression levels for all of the mutants and wild-type yCD, with no detectable expression in vector control pools (data not shown). Pools of stable transfectants were assayed for 5FC sensitivity over a drug range of 1 to 10mM. Representative results of the 5FC sensitivities displayed by the yCD mutants, wild-type yCD and a vector control are shown in Figure 2. Little to no toxicity was observed with vector alone at the lower 5FC doses; however, above 6mM 5FC an inherent cytotoxicity is observable in the glioma cell line. YCD-D92E displays the greatest reduction in IC50 (~30%) for 5FC of the three regio-specific random mutants (Fig. 2a). No difference in sensitivity was observed with the yCD-I98L substitution as compared to wild-type yCD. The yCD-M93L mutant displays an intermediate IC50 of 9.5mM, an estimated decrease of ~15% from wild-type yCD.
Figure 2.
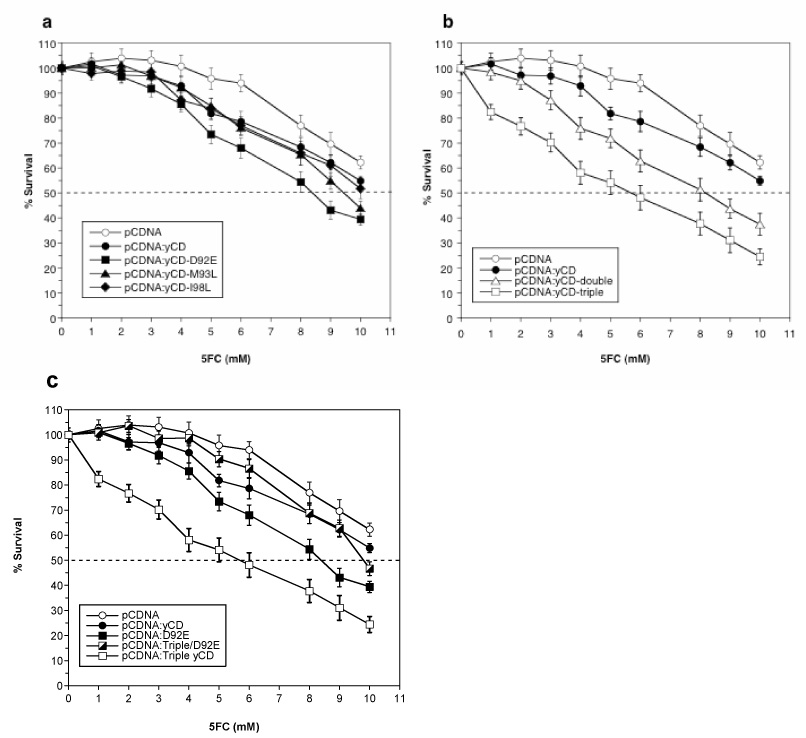
Sensitivity of selected, computationally designed or superimposed mutant yCD-expressing rat C6 glioma transfectants to 5FC. Pools of stable transfectants containing vector only (pCDNA), wild-type yeast cytosine deaminase (yCD), a) regio-specific random mutants (D92E, M93L or I98L), b) computationally designed thermostable mutants (double and triple) or c) the superimposed mutant (yCD-triple/D92E) were used to transfect rat C6 glioma cells and evaluated for 5FC sensitivity as described in Materials and Methods. After six days of 5FC treatment, cell survival was determined using Alamar Blue according to the manufacturer’s instructions. Each data point (mean ± SEM, n=3 performed with at least fifteen replicates) is expressed as a percentage of the value for control wells with no 5FC treatment.
The computationally designed thermostablizing yCD-double and yCD-triple mutants also display increased sensitivity towards 5FC in glioma cells (Fig. 2b). The yCD-double mutant displays a similar IC50 (8mM) to yCD-D92E, an approximate 30% reduction in IC50 compared to wild-type yCD-transfected cells. The greatest enhancement in activity was observed with the yCD-triple mutant with an IC50 of approximately 6mM, an estimated 50% reduction relative to wild-type yCD.
Unexpectedly, none of the superimposed mutants exhibit enhanced activities towards 5FC. The yCD-triple/I98L and yCD-triple/M93L have an IC50 of 9mM (data not shown) while the yCD-triple/D92E has a similar IC50 to that of wild-type yCD transfected cells (Fig. 2c). These results indicate that the addition of the randomly generated substitutions to the designed mutant negates the effect introduced by the triple substitutions.
5FC sensitivity assayed using a xenograft tumor model
The behavior of the two most active yCD constructs from the assays above (yCD-triple and yCD-D92E) was then further compared to wild-type enzyme and a negative control in a mouse xenograft model of prodrug gene therapy. As described in Materials and Methods, pools of rat C6 cells stably transfected with pCDNA containing yCD, yCD-triple, yCD-D92E or vector alone (0.5 × 106 cells) were subcutaneously injected into the flanks of nude mice (n= 5). When the tumor size reached 3–4mm, phosphate buffered saline (PBS) or 5FC at 500mg/kg was injected intraperitoneally once a day for 18 days. Tumor size was monitored every other day. Tumor cells transfected with vector only (pCDNA) showed no statistical difference in tumor size between mice treated with PBS or prodrug (Fig. 3a). Similarly, no statistical difference was observed in tumor volume in mice seeded with cells transfected with pCDNA:yCD that were injected with PBS or 5FC (Fig. 3b). The lack of difference in wild-type yCD tumor size between PBS and 5FC treated mice at day 18 is likely a reflection of the low 5FC dose administered and/or the duration of the prodrug treatment.
Figure 3.
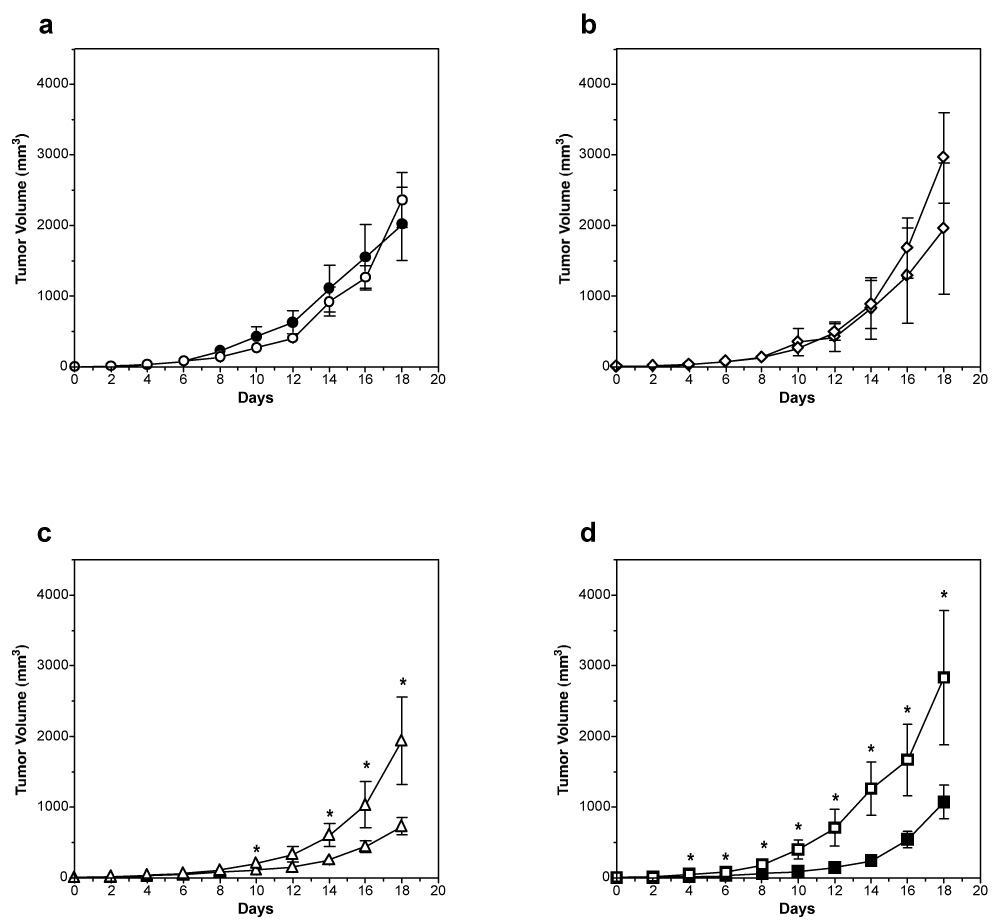
Growth of tumors expressing yCD during 5FC treatment. Pools of rat C6 glioma cells transfected with a) vector, b) wild-type yCD, c) yCD-triple or d) yCD-D92E were used to seed tumors in nude mice. When tumor size reached 3–4mm (day 0), PBS (open symbols) or 5FC (500mg/kg, closed symbols) was administered once a day for 18 days. During this period tumor growth was measured every other day (n = 5 for each group). Tumor volume was calculated using the formula 4/3π ((width × length × height)/2), plotted and analyzed for statistical significance using the Student’s T-test. Asterisks denote statistical significance (P = 0.05 or lower).
In contrast to tumors expressing wild-type yCD, the prodrug treated mice bearing yCD-triple or yCD-D92E expressing tumors elicited a strong response (Fig. 3c and 3d). The mice bearing yCD-triple expressing tumors treated with 5FC displayed the greatest restriction in tumor growth (mean tumor volume of 1076mm3 versus PBS tumor mean of 2833mm3; P = 0.05). The yCD-D92E tumor bearing mice also showed restricted growth in the presence of 5FC (733mm3 versus 1942mm3 for saline treated mice; P = 0.035). To compare the effects of 5FC on tumors expressing the redesigned thermostabilized yCD-triple with that of the selected yCD-D92E, the mean tumor size of the prodrug treated group was divided by the mean tumor size of the prodrug treated group for each group on day 18. Both yCD-triple and yCD-D92E tumor bearing mice responded equally well (2.65 versus 2.64, respectively).
Enzyme kinetics and thermostability
The unexpected loss of in vitro sensitivity towards 5FC when yCD-triple and yCD-D92E were combined, as compared to their individual behaviors, led us to investigate the catalytic and biophysical properties of these yCD variants in more detail. Using purified proteins, enzyme assays with cytosine and 5FC as substrates were performed as described in Materials and Methods.
Overall, the relative catalytic efficiency (kcat /Km) towards cytosine of the individual yCD-triple or yCD-D92E mutants is not appreciably different from wild-type enzyme, whereas the yCD-triple/D92E is approximately 3-fold less efficient (Table 1). This result is primarily caused by a reduction in the enzyme's turnover rate: kcat (cytosine) for yCD-triple/D92E is approximately 1/8th that of wild-type enzyme. In contrast, the Km values for cytosine observed with all mutant enzymes are all within approximately 2 to 2.5-fold of wild-type yCD values (Table 1).
Table 1.
Enzyme kinetics
| Substrate | Km | Vmax | kcat | kcat/Km | |
|---|---|---|---|---|---|
| (mM substrate) | (M prod/sec) | (M prod/sec M E) | (1/sec M E) | ||
| yCD-wild-type | 5FC | 0.083 | 1.87E-05 | 18.8 | 2.26E+05 |
| yCD-triple | 5FC | 0.14 | 2.95E-05 | 29.5 | 2.09E+05 |
| yCD-D92E | 5FC | 0.15 | 3.17E-05 | 31.7 | 2.16E+05 |
| yCD-triple/D92E | 5FC | 0.068 | 9.51E-06 | 9.5 | 1.39E+05 |
| yCD-wild-type | Cytosine | 1.17 | 1.70E-04 | 170 | 1.45E+05 |
| yCD-triple | Cytosine | 0.57 | 8.62E-05 | 86.2 | 1.53E+05 |
| yCD-D92E | Cytosine | 0.52 | 5.18E-05 | 51.8 | 1.00E+05 |
| yCD-triple/D92E | Cytosine | 0.47 | 6.53E-05 | 21.8 | 4.59E+04 |
When the prodrug 5FC is instead used as the substrate in kinetic assays, the individual yCD-triple and yCD-D92E mutant enzymes again display overall catalytic efficiencies (kcat/Km) that are roughly equivalent to wild-type yCD (Table 1). This corresponds to slightly higher Km values (1.7- and 1.8-fold, respectively) balanced by small increases in kcat. In contrast, although the combined yCD-triple/D92E enzyme has a slightly lower Km, it displays a kcat value half that of wild-type yCD. This gives the combined yCD-triple/D92E mutant enzyme a slightly impaired 5FC catalytic efficiency (1.6-fold) compared to wild-type yCD, yCD-triple and yCD-D92E.
Inside a cell, cytosine and 5FC compete for the active site of yCD. To take this into account, the relative specificities or substrate preference for 5FC of all three mutant enzymes was compared (calculated as [kcat/Km (5FC)/(kcat/Km (5FC) + kcat/Km (cyt))]). With wild-type yCD set at a relative specificity of 1.0, there is no considerable difference in substrate preference between yCD-wild-type (1), yCD-triple (0.947) or yCD-D92E (1.12) and only a modest 1.23-fold increase in preference for 5FC displayed by the yCD-triple/D92E mutant. This modest shift in specificity towards 5FC for the combined mutant is not correlated with what was observed in cell sensitivity assays, where yCD-triple/D92E performed no better than the wild-type enzyme.
In previous experiments we showed that the redesigned yCD-double and yCD-triple mutants display unfolding temperatures (Tm) in circular dichroism experiments that are 6°C and 10°C higher, respectively, than that of the wild-type enzyme (wild-type yCD, Tm = 52°C; yCD-double and yCD-triple mutants, Tms= 58°C and 62° C, respectively)30. The stabilization of yCD in the yCD-triple mutant corresponds to improved complementation of CD activity in E. coli at elevated temperatures30, and presumably is also responsible for its improved performance in PGT assays in this study.
We therefore examined the effects of the individual yCD-D92E substitution and of the superimposed yCD-triple/D92E mutation on protein stability. Thermal unfolding experiments were performed on the wild-type, yCD-triple, yCD-D92E and yCD-triple/D92E proteins using circular dichroism spectroscopy as outlined in Materials and Methods. Although the yCD-D92E mutant was selected based on its ability to confer increased sensitivity to 5FC in E. coli, this enzyme unexpectedly also displays an increase in its apparent denaturation temperature, corresponding to a Tm 4°C higher than the Tm for wild-type yCD. Combination of yCD-D92E with the previous yCD-triple mutations in the enzyme core results in a dramatic 16°C increase in apparent Tm as compared to wild-type yCD (6°C higher than that of the yCD-triple mutant). For all of the enzyme constructs, thermal unfolding was irreversible, presumably due to aggregation. Similar to previous studies of the computationally repacked yCD variants30, the half-life of the enzyme activity for yCD-D92E and for yCD-triple/D92E is increased relative to their wild-type and yCD-triple parents, respectively (data not shown).
Structure determination
The crystal structure of yCD-triple/D92E bound to a transition state analogue was solved at 2.3 Å resolution (Table 2) and compared to both previously solved wild-type (PDB 1p6o) and triple mutant (PDB 1ysb) structures in order to identify any potential structural changes that may explain the observed IC50 values as well as the thermodynamic and kinetic data. The yCD-D92E and the yCD-triple mutations appear far from the active site (Fig. 5a) and indeed the active site residues show no perturbations when either the yCD-triple or yCD-D92E/triple structures are compared to wild-type enzyme (Fig. 5b). The yCD-D92E mutation, although close in sequence to active site residues C91 and C94, is actually involved in the dimer interface of the homodimeric enzyme. In the wild-type enzyme both carboxyl oxygens in aspartate 92 make salt-bridge contact to a nearby arginine residue in the adjacent monomer (Fig. 6a), with oxygen to nitrogen distances of 3 to 3.5 Å. In yCD-D92E the extra carbon in the glutamate is packed against a nearby lysine and isoleucine, resulting in a redistribution of contacts made by the carboxylate oxygens of the mutated side chain (Fig. 6b). In this structure one oxygen to nitrogen distance is considerably shorter (approximately 2 Å) while the second is lengthened.
Table 2.
Data collection and refinement statistics
| yCD-triple/D92E Bound to DHP | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions (Å) | 55.9, 69.1, 74.2 |
| Resolution (Å) | 2.30 (2.38-2.30) |
| Rmerge | 0.042 (0.087) |
| I / σI | 8.5 (7.5) |
| Completeness (%) | 99.2 (98.8) |
| Redundancy | 7.2 (7.3) |
| Total reflections | 96421 |
| Unique reflections | 13334 |
| Refinement | |
| Resolution (Å) | 2.30 |
| No. reflections | 13204 |
| Rwork / Rfree | 0.204/0.258 |
| No. atoms | 2548 |
| Protein | 2396 |
| Ligand/ion | 19 |
| Water | 133 |
| B-factors | 18.1 |
| Protein | 17.9 |
| Ligand/ion | 11.3 |
| Water | 24.2 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.10 |
| Ramachandran Distribution | 98.7, 1.0, 0.3 |
| (% core, allowed, disallowed) |
Number of xtals for each structure should be noted in footnote.
Values in parentheses are for highest-resolution shell.
Figure 5.
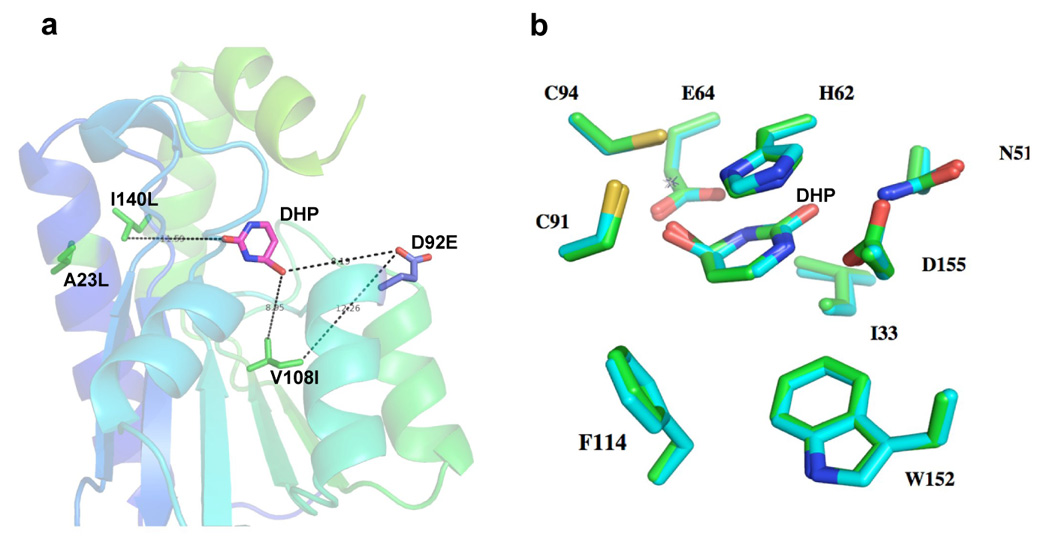
Crystal structure analysis of the yCD-triple/D92E active site. a) Location and relative distances of mutated residues from a mechanism-based inhibitor (dihydropyrmidine or DHP) that acts as a mimic of the reaction transition-state. All residue atoms are located over 8 Å from the substrate analogue, including the residue (D92E) selected in a screen for 5FC sensitization. b) Superimposition of wild-type and yCD-triple/D92E active site residues.
Figure 6.
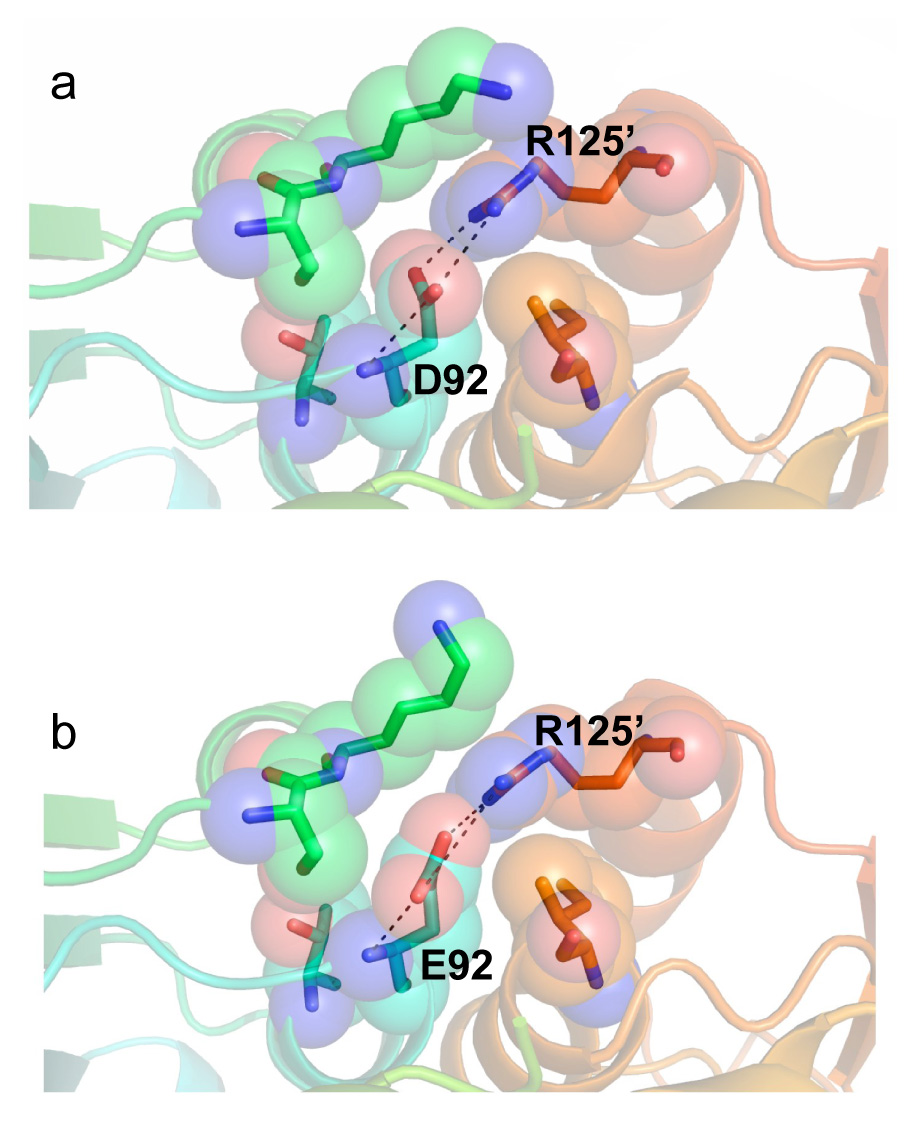
Interaction and position of (a) wild-type D92 at the dimer interface and (b) mutant E92 at the dimer interface with its surrounding neighbors. The mutation causes a very small rearrangement of the side chain torsion angles and interactions between the mutated residue and its nearest neighbor, R125. No other significant structural perturbations due to D92E are apparent.
DISCUSSION
The initial premise of this study was that independent mutations in the active site and the hydrophobic core of yCD, that respectively influence substrate specificity and enzyme thermostability, could each be identified in separate experiments and then combined in a single construct for maximum benefit. Furthermore, whereas stabilizing interactions in a protein core can be improved through computational protein design30, an enzyme's substrate specificity is still more effectively addressed through incorporation of random mutations and subsequent genetic screens or directed evolution protocols. Thus, a two-pronged experimental approach to optimize the physical and catalytic properties of yCD was expected to yield a combination of enzyme mutations with optimal properties.
Instead, the experiments described above yielded unexpected results in three ways. First, the protocol of random mutagenesis and genetic screening generated a construct (yCD-D92E) that displayed an increase in its unfolding temperature Tm, in addition to a small (1.12-fold) specificity shift towards 5FC relative to cytosine. This result appears to have been made possible through the choice of residues that were subjected to mutation, which extended beyond the substrate binding pocket, to nearby positions involved in dimerization. Residues were chosen for mutation prior to determination of the enzyme's structure, based on sequence conservation across the closest known homologues with deaminase function. In retrospect, strongly conserved residues in yCD (and most other enzymes) include not only those positions most critically involved in substrate recognition and catalysis, but also amino acids those that closely couple the architecture of the active site to the fold and stability of the enzyme tertiary structure (such as D92).
Second, the stabilized mutant yCD constructs produced by repacking of the hydrophobic core (yCD-double and yCD-triple) were significantly improved in their performance in PGT assays, with the latter construct actually outperforming the best mutant construct selected solely on the basis of 5FC sensitivity. This result, considered in concert with the properties of yCD-D92E described above (improved stability and nominal changes in catalytic behavior), may indicate that the primary limiting behavior of wild-type yCD for prodrug gene therapy is its stability, which presumably is coupled to steady-state enzyme levels in transfected cells and enhanced conversion of 5FC to 5FU.
Finally, the combination of two sets of physically separate mutations (yCD-triple and yCD-D92E) that were independently generated failed to act in a synergistic or additive manner in PGT assays when combined into a single construct. On the surface, an explanation for this behavior seems straightforward: the combined construct (yCD-triple/D92E) is significantly more thermostable than are any of the individual yCD constructs (with an apparent increase in Tm of 16°C), which might lead to a corresponding reduction in catalytic efficiency at physiological temperatures due to reduced conformational dynamic flexibility of the protein backbone. However, the difference in kinetic rate and catalytic efficiency towards 5FC exhibited by yCD-triple or yCD-D92E compared to yCD-triple/D92E (which performs similarly to wild-type enzyme) is not dramatic: the combined mutant is perhaps 1.5-fold less active towards the prodrug than are the individual mutants. This result might imply that there exists a quite sharp 'threshold' of activity towards 5FC deamination in the cell that leads to considerably more sensitivity towards low concentrations of the prodrug. For this to be true, the specific activity of the wild-type enzyme under standard protocols of transfection and expression would have to be somewhat close to that critical point of metabolic sensitivity. However, alternative situations might also explain the poor performance of the combined mutations in PGT assays, including (but not limited to) unexpectedly reduced expression levels in eukaryotic cells, abnormal protein localization, trafficking, or protein turnover, or unexpected effects on kinetic specificity and activity (for example, inhibition in cells by a nucleobase analogue that is not accounted for in these studies).
Variable routes to protein thermostabilization
Two entirely separate forms of thermostabilized yCD enzyme have been generated in these studies, one of which was generated in a screen for increased sensitivity to 5FC, the other in a structure-based redesign protocol intended to produce a more robust form of the enzyme. The independent sequence changes that lead to stabilization (increased van der Waals packing of each subunit's hydrophobic core, versus more subtle alteration of contacts in the homodimer interface) are structurally dissimilar from one another, providing a clear demonstration that multiple evolutionary routes can act on protein stability through considerably different structural mechanisms.
This result reinforces a myriad of studies of protein stability conducted over the past ten years, where the structures of homologous proteins from various biological sources (ranging from cryophiles to mesophiles to thermophiles) have been directly compared in attempts to catalogue the determinants of protein stability31–36. Although a variety of factors have been implicated in thermostability (including hydrophobic packing, formation of additional salt-bridges within the protein structure, modifications of surface charge distribution and shifts in amino acid composition, to name just a few), each mechanism appears to be relatively unique to specific structural families of proteins. In addition, individual proteins can be made thermostable either by reducing their relative free energy in the folded state (termed 'thermodynamic stabilization') or by decreasing the rate of unfolding through modifications that increase the energy barrier of the unfolding transition state (termed 'kinetic stabilization'). This latter mechanism for protein stabilization has been observed for a variety of proteins, such as α-lytic protease37. A significant aspect of such proteins is the identification of residues at points distant from the hydrophobic core, and often at domain interfaces, that are highly sensitive to mutational effects on protein stability38. Given the nature of the yCD-D92E mutation at the dimer interface, which does not appear to obviously increase favorable contacts in the folded enzyme, it might be possible that this mutation stabilizes yCD via this latter mechanism.
yCD and 5FC and enzyme engineering in recent gene therapy trials and the role of optimized enzyme constructs
Between 1990 and 2004, 405 gene therapy trials were approved in the US for cancer treatment, of which 95 contain various elements of enzyme/prodrug therapeutic strategies39; 78 of these trials were active as of July 2007 (http://www.wiley.co.uk/genetherapy/clinical/). Three of these trials, all involving the combination of thymidine kinase and ganciclovir, have advanced to phase III multicenter programs39–40. A large percentage of these trials have targeted prostate cancer, due to the relatively low percentage of these cancers that have associated detectable metastatic disease (<10%) at the time of diagnosis11. Of these latter trials, many have made use of cytosine deaminase enzymes and 5FC, most in combination with additional enzyme/prodrug activities11–15. Results from phase I trials against prostate cancer and pancreatic cancer have recently been reported for combined PGT therapies involving yeast cytosine deaminase. In each study a replication-competent adenovirus armed with wild-type yCD fused to a modified HSV-thymidine kinase (SR39), in combination with 5FC/ganciclovir prodrug administration and radiation, was utilized11. In both cases, the trimodal approach (oncolytic virus, suicide gene/prodrug, and radiation) significantly increased tumor control beyond that displayed by individual modalities, with a corresponding reduction in morbidity and toxicity as compared to traditional chemotherapy. As well, positron emission tomography (PET) studies indicated strict localization of the transfected gene and its product in the targeted tissue.
Despite the promise of such studies, prodrug gene therapy is limited pharmacokinetically by the ability to deliver the suicide gene efficiently to tumor cells whereas traditional chemotherapeutic strategies are perhaps equally limited by the ability to specifically target the effects of cytotoxic agents to tumor cells at levels over normal proliferating cell populations in the body. The ability of PGT to effectively ablate tumor cells is also reliant on the inherent sensitivity that cell type displays towards a particular gene/prodrug combination. Additionally, when poor responsiveness necessitates repeat therapeutic intervention, rapid immune clearance of cells expressing a previously seen foreign protein may serve to further limit treatment efficacy. As such, various enzyme/prodrug combinations will be required to accommodate these various treatment scenarios. To offset the significant hurdles associated with PGT, the pharmacokinetic properties of the enzyme (its stability, half-life and kinetic activity), the prodrug (its metabolism and toxicity) and combination of the two (their uniqueness to transfected cells) must be optimized for maximum therapeutic efficacy. In particular, the catalytic efficiency of enzymatic activation of the prodrug, and the relative level of activation of the prodrug in transfected cells must be optimized for several enzyme/prodrug combinations. For the most commonly used enzyme/prodrug combination under clinical study to date (HSV thymidine kinase and ganciclovir), random mutagenesis and genetic selection experiments have generated a variant of HSV thymidine kinase (SR39), that has led to a 294-fold increase in sensitivity to ganciclovir when compared to wild-type enzyme, as well as improved tumor growth inhibition41–42.
In the case of cytosine deaminase and 5FC, the availability of two separate enzyme scaffolds and folds (yCD and bCD) further increases the parameters that can be optimized and combined with other enzymes; each enzyme displays unique characteristics of size, oligomeric structure, behavior when 'tethered' to additional enzymes, metal dependence, stability and substrate specificity profiles. Given that a variety of studies that use two enzyme/prodrug combinations have shown synergistic improvements in efficacy over individual enzyme systems43–46, the utility of kinetic and biophysical optimization of each enzyme seems clear. Thus, side-by-side optimization of each enzyme source for PGT is likely to illuminate the balance of properties that are most critical for performance in gene therapy applications.
MATERIALS AND METHODS
Materials
Oligonucleotides used to mutate and sequence yCD were obtained from either Sigma-Proligo (St. Louis, MO) or Integrated DNA Technologies (Coralville, IA). Restriction enzymes were obtained from New England Biolabs (Beverly, MA). The plasmid pETHT:yCD expressing His-tagged yCD was constructed as described in Ireton et al3. DNA purification was done using several kits (Wizard PCR prep kits Promega (Madison, WI), HiSpeed Plasmid Mini Kit from Qiagen (Valencia, CA) and StrataPrep EF Plasmid Midikit from Stratagene (La Jolla, CA)). Alamar Blue was purchased from Serotec Limited (Oxford, UK). All cell culture reagents were purchased from Gibco (Carlsbad, CA). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Bacterial strains
Escherichia coli GIA39, a strain deficient in cytosine deaminase and orotidine 5’- phosphate decarboxylase, was obtained from the E. coli Genetic Stock Center (CGSC #5594: th− dadB3 fhuA21 codA1 lacY1 tsk-95 glnV44(AS) λ− pyrF101 his-108 argG6 ilvA634 thi-1 deoC1 glt-15). E. coli GIA39 was lysogenized with DE3 according to the manufacturer’s instructions (Novagen, Madison, WI). The derived strain, GIA39(DE3), was used in the genetic complementation assays for cytosine deaminase activity. E. coli strain CJ236 (F+LAM−, ung-1, relA1, dut-1, spoT1, thi-1) was used to produce single-stranded DNA for site-directed mutagenesis procedures. The E. coli strain NM522 (F+ lacIqΔ(lacZ)-M15 proA+B+/supE thiΔ(lac-proAB) Δ(hsdMS-mcrB)5(rk−mk−McrBC−)) and E. coli strain XL1-Blue (F'::Tn10 proA+B+ laclqD(lacZ) M15/recA1 endA1 gyrA96 (Nalr) thi hsdR17 (rk−mk+) supE44 relA1 lac) were used as recipients for certain cloning procedures. E. coli BL21-RIL (Novagen, Madison, WI) was used for large scale protein purification.
Construction of the yCD regio-specific random library
Preparation of the randomized insert
The basic protocol for regio-specific random mutagenesis was performed as outlined in Kurtz and Black47. Table 3 lists the oligonucleotides MB223-MB236 designed to synthesize a 139bp dsDNA fragment that spans 12 codons (T83, L84, Y85, T86, L88, S89, D92, M93, T95, G96 and I98) and contains 21% randomness at those positions.
Table 3.
List of oligonucleotides
| Oligonucleotides used to generate the randomized DNA fragment. |
| MB223 (56mer), 5’ GTGAGATCTCCACTTTGGAAAACTGTGGGAGATTAGAGGGCAAA GTGTACAAAGAT 3’ |
| MB224 (95mer), 5’ CCGACAA(CACAGCGTG)GAATACCATACATGATGATGGCACCTGTACACATGTCGCATGG AGACAGCGTCGTATACAAAGTGGTATCTTTGTACAC 3’ |
| MB225 (18mer), 5’ GTG(AGATCT)CCACTTTGG 3’ |
| MB226 (17mer), 5’ CCGACAACACAGCGTGG 3’ |
| Nucleotides show in bold were mixtures of wild-type nucleotide (79%) and the three non-wild-type nucleotides (21%). Parenthesis denote DraIII (MB224) and Bg/II (MB225) restriction sites used for cloning purposes. |
| Oligonucleotides used to introduce or eliminate restriction sites. |
| MB277 (21mer), 5’ CGATGGCCCAATACGTGAACC 3’ |
| MB307 (22mer), 5’ CGGGATCGCGATCGCGGGCAGC 3’ |
| MB374 (31mer), 5’ CGCTGTCTCCATGCGAAATGTGTACAGGTGC 3’ |
| MB375 (31mer), 5’ CGACCTGTACACATTTCGCATGGAGACAGCG 3’ |
| MB376 (31mer), 5’ CGCTGTCTCCATGCGACCTGTGTACAGGTGC 3’ |
| MB377 (31mer), 5’ GCACCTGTACACAGGTCGCATGGAGACAGCG 3’ |
| MB378 (34 mer), 5’ GCGACATGTGTACAGGAGCCCTCATCATGTATGG 3’ |
| MB379 (34 mer), 5’ CCATACATGATGAGGGCTCCTGTACACATGTCGC 3’ |
| Nucleotides shown in bold were altered. Underlined nucleotides indicate change in restriction site. |
Briefly, the 139bp dsDNA fragment was synthesized by annealing 50pmol each of MB223 and MB224 with 10 × annealing buffer (70mM Tris-HCl at pH 7.5, 60mM MgCl2, 200mM NaCl) in a final volume of 50µL at 95°C for 5min, 65°C for 20min, and room temperature for 10min. Next, the annealed product was extended with the Klenow fragment of E. coli DNA polymerase in an 80µL reaction mixture consisting of the 40µL annealed product, 4µL 10 × annealing buffer, 5.6µL 10mM dNTPs, 1.6µL 0.1M DTT and 4.8µL Klenow (5U/µL) at 37°C for 30min, 65°C for 10min and room temperature for 10min.
Amplification of the random insert
A master mix was prepared consisting of 110µL 10 × PCR buffer (200mM Tris-HCl at pH 8.3, 250mM KCl, 15mM MgCl2, 0.5% Tween-20), 200pmol MB225 and MB226 each, 4.4µL 10mg/mL BSA, 5.5µL 10mM dNTPs and 4.4µL Taq polymerase (5U/µL). The extended product (16pmol or 5µL) and 27.8µL of the master mix were mixed to a final volume of 200µL and split into four tubes containing 50µL each. The 50µL mixtures were subjected to amplification using an Eppendorf Thermocycler by 30 cycles with 1 cycle at 94°C for 1min, 34°C for 2min, and followed with a 7min extension at 72°C. Amplification of the 139bp insert was confirmed by gel electrophoresis.
Construction of recombinant yCD variants
To construct the vector carrying the inactive or dummy gene, the DraIII and Bg/II sites in the vector backbone were removed by site-directed mutagenesis48 using the oligonucleotides MB277, to remove the DraIII site, and MB307, to remove the Bg/II site (Table 3). To reduce the presence of wild-type yCD in the selection, the yCD gene was inactivated by restriction with AccI followed by extension with the Klenow fragment and religation. The resulting inactivated yCD was designated as “pETHT:yCD-dummy.” Following digestion with Bg/II and DraIII, the gel purified insert was cloned into the vector digested with the same restriction enzymes.
Transformation of E. coli GIA39(DE3) and positive selection
Approximately 3.5µL of the ligated product was electroporated into 40µL of electrocompetent E. coli GIA39(DE3) and then shaken at 37°C for 1hr in 1mL of SOC medium (3g Bactopeptone, 2.5g yeast extract, 1M NaCl, 1M KCl, 5mM MgSO4, 5mM MgCl2 and 1.8% glucose per liter). The transformation mixture was concentrated by pelleting, resuspended in 100µL of 0.9% NaCl and plated at various volumes onto 2 × YT rich medium, uracil and cytosine minimal media plates supplemented with 50µg/mL carbenicillin. Growth on cytosine minimal medium requires the presence of a functional yCD, while 2 × YT and uracil minimal media were used as positive controls. Uracil minimal medium (500mL) was prepared from 0.36g yeast synthetic dropout without leucine, 50mL 10 × M9 salts (15g KH2PO4, 33.9g anhydrous NaHPO4, 2.5g NaCl, 5.0g NH4Cl), 1mM MgSO4, 2.5mL, 20% glucose, 0.1mM CaCl2, 1mL 2% leucine, and 7.5g Bactoagar. Cytosine minimal medium (500mL) was prepared from 1mM cytosine, 0.96g yeast synthetic dropout without uracil, 8.5g Bactoagar, 50mL 10 × M9 salts, 1mM MgSO4, 2.5mL 20% glucose, and 0.1mM CaCl2. The 2 × YT plates were incubated at 37°C overnight, and the uracil and cytosine plates were incubated at 37°C for approximately 36hr. The number of transformants on the 2 × YT plate allows estimation of the library size. Transformants from the cytosine plates were picked and restreaked onto fresh cytosine plates to confirm the phenotype.
Selection of 5FC sensitive mutants
Genetic complementation of yCD was done as previously established for bacterial CD49. To determine the ability of the mutants to confer 5FC sensitivity, the functional variants determined by the positive selection described above were streaked onto cytosine plates supplemented with 5FC at 10µg/mL, a sublethal dose for wild-type yCD, and incubated for approximately 36hr at 37°C. Colonies unable to grow on the 5FC plates were selected from the control plates and subjected to additional rounds of negative selection at decreasing 5FC concentrations (5, 2, 1 and 0.5µg/mL). Plasmid DNA of the yCD variants was isolated and the randomized region sequenced using the T7 terminator primer (5’TATGCTAGTTATTGCTCAG 3’) at the core sequencing laboratory at Washington State University.
Construction of mammalian expression vectors
Computationally designed thermostabilized yCD genes described in Korkegian et al.30 (pET15b:yCD-double and pET15b:yCD-triple) were sub-cloned into the mammalian expression vector, pCDNA6/myc-His B (Invitrogen, Carlsbad, CA). The yCD-double and yCD-triple genes were subcloned into pCDNA6/myc-His B digested with EcoRV and XhoI as NcoI (blunt-ended)/XhoI fragments. After restriction enzyme verification, DNA sequencing analysis confirmed the presence of the yCD-double and yCD-triple genes.
Site-directed mutagenesis was performed to introduce the mutations derived from the regio-specific random mutagenesis to the yCD-triple mutant using the QuikChange Site-directed Mutagenesis Kit from Stratagene (La Jolla, CA) according to the manufacturer’s protocol. Three pairs of oligonucleotides containing the D92E9 (MB374/MB375-Af/III), M93L (MB376/MB377-Af/III) or I98L (MB378/MB379-BanI) substitutions and a silent mutation to remove a restriction site for screening purposes were synthesized by Integrated DNA Technologies (Coralville, IA)(Table 3). After restriction enzyme verification, DNA sequencing analysis was performed to confirm the presence of the correct mutation.
5FC sensitivity assays
One µg of each DNA (pCDNA (vector alone), pCDNA:yCD, pCDNA:yCD-D92E, pCDNA:yCD-M93L, pCDNA:yCD-I98L, pCDNA:yCD-double, pCDNA:yCD-triple;, pCDNA:yCD-triple/D92E; pCDNA:yCD-triple/M93L; pCDNA:yCD-triple/I98L) was used to transfect 1 × 105 rat C6 glioma cells by lipofection using FuGENE 6 transfection reagent (Roche Diagnostics, Penzberg, Germany) at a 3:1 ratio according to the manufacturer’s directions. Immunoblots were performed to assess protein expression. Briefly, pools of transfectants were harvested and resuspended at 100,000 cells/µL in lysis buffer (for 2mL: 2µL 1M DTT, 20µL 1M HEPES, 40µL Nonidet P40 (Roche Diagnostics, Pernzberg, Germany) 2µL MgAc2, H2O to final volume). The resuspended pellets were incubated on ice for 20min and subjected to centrifugation at 4°C for 20min to pellet debris. Heat denatured samples (10µL per well) were subjected electrophoresis on a 15% SDS containing polyacrylamide gel, transferred to a nitrocellulose membrane and blocked with 3% gelatin in Tris-buffered saline. The membrane was probed with rabbit polyclonal yCD antiserum (gift from Dr. Alnawaz Rehemtulla, U. Michigan, Ann Arbor, MI) followed by goat anti-rabbit AP-conjugated antiserum. The blot was developed using the AP Conjugate Substrate Kit (Bio-Rad, Hercules, CA). To determine the cytotoxicity of 5FC, pools of transfectants were transferred to 96 well microtiter plates at an initial density of 250 cells per well in DMEM plus supplements50. After cell adherence overnight, 5FC (0- 10mM) was added in sets of eight wells for each concentration tested. The plates were incubated for 6 days at 37°C in 5% CO2 at which time the redox-indicator dye Alamar Blue was added. Cell survival was determined several hours later as according to the manufacturer’s instructions and the data were plotted with a standard error of the mean bar. At least three replicates were performed.
Xenograft tumor model
Pools of C6 glioma cells stably transfected with pCDNA, pCDNA:yCD, pCDNA:yCD-D92E, or pCDNA:yCD-triple (0.5 × 106 cells in 200µL of phosphate buffer saline (PBS) at pH 7.2) were injected subcutaneously into 5- to 6- week-old female nude mice (n = 5 for each group) (BALB/cAnNCr-nu/nu; National Cancer Institute, Frederick, MD, USA). When the tumors reached 3–4mm (day 0), PBS or 5FC (500mg/kg) was administered by intraperitoneal injection once a day for 18 consecutive days. Starting at day 0, the tumor volume was monitored using caliper measurement (length, width, and height) every other day. Tumor volume was calculated using the formula: 4/3π((widthxlengthxheight)/2). Tumor volume was plotted and analyzed for statistical significance using Student’s T-test.
Protein expression and purification
Protein expression and purification of yCD was carried out as previously described3, with the exception that buffer exchange was carried out with Biorad pre-packed Econo-Pac 10DG buffer exchange columns rather than overnight dialysis. Protein expression from pET15b:yCD (BL21-RIL) yields yCD with a thrombin cleavable 6- His tag fused to the N-terminus. All kinetics and thermostability experiments were carried out with fresh, unfrozen protein stored at 4°C for 2 weeks or less.
Activity assays
The conversion of cytosine to uracil by yCD was measured spectrophotometrically by monitoring change in absorbance at 286nm while the conversion of 5FC to 5FU was monitored at 238nm30,49. The protein was diluted to 2µM in 50mM Tris-Cl (pH 7.5) and mixed 1:1 with a range of nine cytosine concentrations from 0.2–1mM in the same buffer. Absorbance at 238nm was measured every 5sec until baseline was reached with the first reading taken 5sec after mixing. Measurements were taken in quadruplicate and averaged to reduce error. Initial velocity was calculated as a function of the initial slope by curve-fitting the resulting plot, taking the derivative and extrapolating back to time zero. Km and kcat values of wild-type yCD and mutant constructs were determined from a double reciprocal (Lineweaver-Burk) plot of the resulting data and the catalytic efficiency kcat/Km was calculated from these values.
Circular dichroism measurement and thermal denaturation experiments
Circular dichroism data were collected on an Aviv 62A DS spectrometer as described in Dantas. et al51. Wavelength scans were run from 200–260nm to determine the folded state of the protein, the ratio of concentration to signal strength and the wavelength where signal strength was at its highest. Temperature melts were run with 8–12µM protein in a 2mm pathlength cuvette from 10° to 98°C in 2° increments with temperature regulated by a Peltier device. Sample temperature was allowed to equilibrate for 30sec before measurement and signal was collected and averaged over 30sec. Denaturation was recorded as a change in elipticity over temperature. Apparent Tms were determined by curve-fitting.
Crystallization and structure determination
Crystals were grown using methodology described in Ireton et al.3. After two days crystals appeared in the drops, but were often twinned. These crystals were then used to streak seed into the clear lower concentration drops where higher quality crystals eventually grew. Crystals were looped out and soaked for 20min in a mother liquor solution containing 2-hydroxypyrimidine concentrated 1.2:1 relative to protein. After soaking the crystals were immediately transferred briefly to a cryobuffer containing the 2- hydroxypyrimidine mother liquor plus 25% DMSO and then flash-frozen in liquid nitrogen. All data were collected from a single crystal on the 5.0.1 beamline at the ALS syncrotron to 1.7 Å. Data were indexed and scaled using the DENZO/SCALEPACK program suite52. Rmerge values for the higher resolution scales were poor so the data was re-indexed and scaled at 2.3 Å. The initial structures were generated by molecular replacement using the program EPMR53 using the wild-type yCD crystal structure with the mutated residues and any residues within a 4 Å shell of those residues truncated to alanine. Refinement was carried out in rounds using the program CNS54–55 with a random 10% of data withheld for cross-validation. The yCD-triple/D92E mutant crystal structure was refined at 2.3Å to a Rwork/Rfree of 20.4/25.7. Electron density maps and models were visualized in XtalView56 while cartoon representations of the structure were generated using PyMOL (DeLano, W. L. (2002) The PyMOL molecular graphics system on World Wide Web. http://www.pymol.org). Coordinates and diffraction data for yCD-triple/D92E structure have been deposited in the PDB database (PDB 2o3k).
Figure 4.
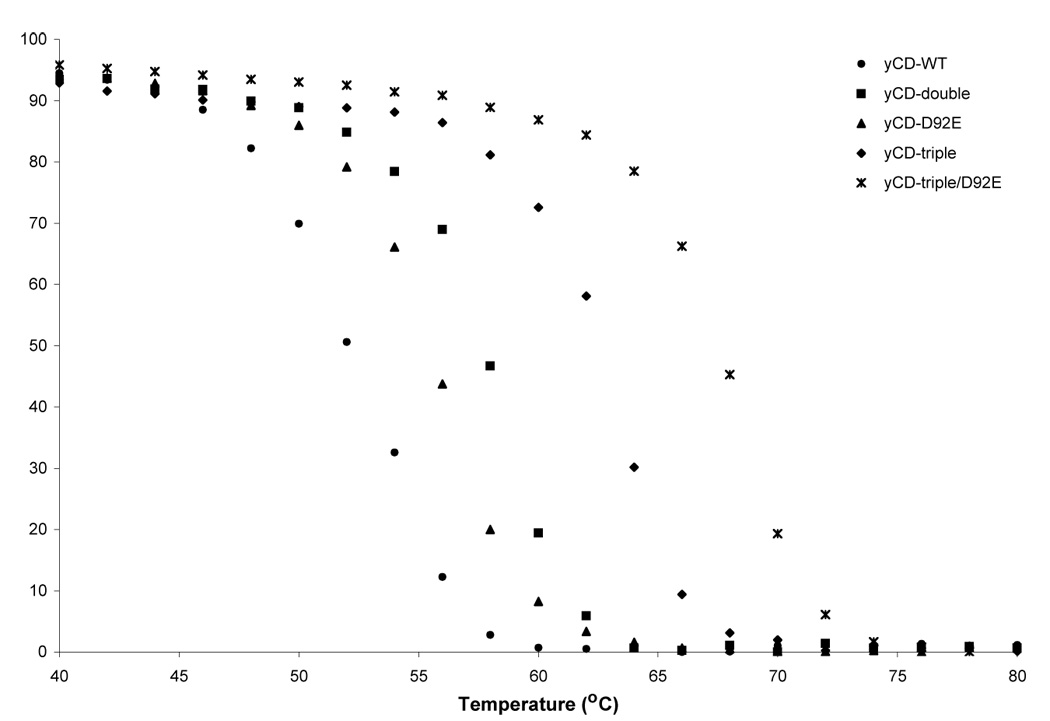
Thermal denaturation measurements of the wild-type and variant yCDs. The temperature melt measures the change in signal at 220nm over a range of temperatures (°C). All constructs show a folded baseline followed by a sigmoidal two-state transition to an unfolded baseline. The baseline plateaus correspond to an assignment of 100% folded protein.
ACKNOWLEDGEMENTS
We thank Dr. A. Rehemtulla for his gift of yCD antiserum. This work was supported by NIH CA85939 (to M.E.B.), NIH CA97328 (to B.L.S. and M.E.B.) and T32-GM08268 (A.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nishiyama T, Kawamura Y, Kawamoto K, Matsumura H, Yamamoto N, Ito T, Ohyama A, Katsuragi T, Sakai T. Antineoplastic effects in rats of 5-fluorocytosine in combination with cytosine deaminase capsules. Cancer Res. 1985;45:1753–1761. [PubMed] [Google Scholar]
- 2.Ireton G, McDermitt G, Black ME, Stoddard BL. The structure of Escherichia coli cytosine deaminase. J. Mol. Biol. 2002;315(4):687–697. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- 3.Ireton GC, Black ME, Stoddard BL. The 1.14 Å crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure (Camb) 2003;11:961–972. doi: 10.1016/s0969-2126(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 4.Huber BE, Richards CA, Krenitsky TA. Retroviral-mediated gene therapy for the treatment of hepatocellular carcinoma: an innovative approach for cancer therapy. Proc. Natl. Acad. Sci. U. S. A. 1991;88:8039–8043. doi: 10.1073/pnas.88.18.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanamaru R, Kakuta H, Sato T, Ishioka C, Wakui A. The inhibitory effects of 5-fluorouracil on the metabolism of preribosomal and ribosomal RNA in L-1210 cells in vitro. Cancer Chemother. Pharmacol. 1986;17:43–46. doi: 10.1007/BF00299864. [DOI] [PubMed] [Google Scholar]
- 6.Pinedo HM, Peters GF. Fluorouracil: biochemistry and pharmacology. J. Clin. Oncol. 1988;6:1653–1664. doi: 10.1200/JCO.1988.6.10.1653. [DOI] [PubMed] [Google Scholar]
- 7.Thomas DM, Zalcberg JR. 5-fluorouracil: a pharmacological paradigm in the use of cytotoxics. Clin. Exp. Pharmacol. Physiol. 1998;25:887–895. doi: 10.1111/j.1440-1681.1998.tb02339.x. [DOI] [PubMed] [Google Scholar]
- 8.Kaliberov SA, Market JM, Gillespie GY, Krendelchtchikova V, Della Manna D, Sellers JC, Kaliberova LN, Black ME, Buchsbaum DJ. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007;14(14):1111–1119. doi: 10.1038/sj.gt.3302965. [DOI] [PubMed] [Google Scholar]
- 9.Hiraoka K, Kimura T, Logg CR, Tai CK, Haga K, Lawson GW, Kasahara N. Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 2007;67(11):5345–5353. doi: 10.1158/0008-5472.CAN-06-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Ye T, Maynard J, Akbulut H, Deisseroth A. Engineering conditionally replication-competent adenoviral vectors carrying the cytosine deaminase gene increases the infectivity and therapeutic effect for breast cancer gene therapy. Cancer Gene Ther. 2006;13(4):346–356. doi: 10.1038/sj.cgt.7700906. [DOI] [PubMed] [Google Scholar]
- 11.Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J, Zhang Y, Barton KN, Brown SL, Lu M, Savera A, Kim JH. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol. Ther. 2007;15(5):1016–1023. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- 12.Post DE, Shim H, Toussaint-Smith E, Van Meir EG. Cancer Scene Investigation: how a cold virus became a tumor killer. Future Oncol. 2005;1:247–258. doi: 10.1517/14796694.1.2.247. [DOI] [PubMed] [Google Scholar]
- 13.Barton KN, Paielli D, Zhang Y, Koul S, Brown SL, Lu M, Seely J, Kim JH, Freytag SO. Second-generation replication-competent oncolytic adenovirus armed with improved suicide genes and ADP gene demonstrates greater efficacy without increased toxicity. Mol Ther. 2006;13(2):347–356. doi: 10.1016/j.ymthe.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Freytag SO, Stricker H, Pegg J, Paielli D, Pradhan DG, Peabody J, DePeralta-Venturina M, Xia X, Brown S, Lu M, Kim JH. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63(21):7497–7506. [PubMed] [Google Scholar]
- 15.Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M, Nafziger D, Pegg J, Paielli D, Brown S, Barton K, Lu M, Aguilar-Cordova E, Kim JH. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002;62(17):4968–4976. [PubMed] [Google Scholar]
- 16.Diasio RB, Laking DE, Bennett JE. Evidence for conversion of 5-fluorocytosine to 5-fluorouracil in humans: Possible factor in 5-Fluorocytosine clinical toxicity. Antimicrob. Agents Chemother. 1978;14:903–908. doi: 10.1128/aac.14.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence TS, Rehemtulla A, Ng EY, Wilson M, Trosko JE, Stetson PL. Preferential cytotoxicity of cells transduced with cytosine deaminase compared to bystander cells after treatment with 5-flucytosine. Cancer Res. 1998;58:2588–2593. [PubMed] [Google Scholar]
- 18.Hoganson DK, Batra RK, Olsen JC, Boucher RC. Comparison of the effects of three different toxin genes and their levels of expression on cell growth and bystander effect in lung adenocarcinoma. Cancer Res. 1996;56:1315–1323. [PubMed] [Google Scholar]
- 19.Kuriyama S, Masui K, Sakamoto T, Nakatani T, Kikukawa M, Tsujinoue H, Mitoro A, Yamazaki M, Yoshiji H, Fukui H, Ikenaka K, Mullen CA, Tsujii T. Bystander effect caused by cytosine deaminase gene and 5-fluorocytosine in vitro is substantially mediated by generated 5-fluorouracil. Anticancer Res. 1998;18:3399–3406. [PubMed] [Google Scholar]
- 20.Nishihara E, Nagayama Y, Narimatsu M, Namba H, Watanabe M, Niwa M, Yamashita S. Treatment of thyroid carcinoma cells with four different suicide gene/prodrug combinations in vitro. Anticancer Res. 1998;18:1521–1525. [PubMed] [Google Scholar]
- 21.Trinh QT, Austin EA, Murray DM, Knick VC, Huber BE. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55:4808–4812. [PubMed] [Google Scholar]
- 22.Kievit E, Nyati MK, Ng E, Stegman LD, Parsels J, Ross BD, Rehemtulla A, Lawrence TS. Yeast cytosine deaminase improves radiosensitization and bystander effect by 5-fluorocytosine of human colorectal cancer xenografts. Cancer Res. 2000;60:6649–6655. [PubMed] [Google Scholar]
- 23.Lawrence TS, Davis MA, Maybaum J. Dependence of 5-fluorouracil-mediated radiosensitization on DNA-directed effects. Int J. Radiat. Oncol. Biol. Phys. 1994;29:519–523. doi: 10.1016/0360-3016(94)90448-0. [DOI] [PubMed] [Google Scholar]
- 24.Hamstra DA, Rice DJ, Pu A, Oyedijo D, Ross BD, Rehemtulla A. Combined radiation and enzyme/prodrug treatment for head and neck cancer in an orthotopic animal model. Radiat. Res. 1999;152:499–507. [PubMed] [Google Scholar]
- 25.Kaliberov SA, Market JM, Gillespie GY, Krendelchtchikova V, Della Manna D, Sellers JC, Kaliberova LN, Black ME, Buchsbaum DJ. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007;14:1111–1119. doi: 10.1038/sj.gt.3302965. [DOI] [PubMed] [Google Scholar]
- 26.Stackhouse MA, Pederson LC, Grizzle WE, Curiel DT, Gebert J, Haack K, Vickers SM, Mayo MS, Buchsbaum DJ. Fractionated radiation therapy in combination with adenoviral delivery of the cytosine deaminase gene and 5-fluorocytosine enhances cytotoxic and antitumor effects in human colorectal and cholangiocarcinoma models. Gene Ther. 2000;7:1019–1026. doi: 10.1038/sj.gt.3301196. [DOI] [PubMed] [Google Scholar]
- 27.Yao L, Li Y, Wu Y, Liu A, Yan H. Product release is rate-limiting in the activation of the prodrug 5-fluorocytosine by yeast cytosine deaminase. Biochem. 2005;44:5940–5947. doi: 10.1021/bi050095n. [DOI] [PubMed] [Google Scholar]
- 28.Kievit E, Bershad E, Ng E, Sethna P, Dev I, Lawrence TS, Rehemtulla A. Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 1999;59:1417–1421. [PubMed] [Google Scholar]
- 29.Katsuragi T, Sakai T, Tonomura K. Implantable enzyme capsules for cancer chemotherapy from bakers' yeast cytosine deaminase immobilized on epoxyacrylic resin and urethane prepolymer. Appl. Biochem. Biotechnol. 1987;16:61–69. doi: 10.1007/BF02798356. [DOI] [PubMed] [Google Scholar]
- 30.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies GJ, Gamblin SJ, Littlechild JA, Watson HC. The structure of a thermally stable 3-phosphoglycerate kinase and a comparison with its mesophilic equivalent. Proteins. 1993;15:283–289. doi: 10.1002/prot.340150306. [DOI] [PubMed] [Google Scholar]
- 32.Yip KS, Stillman TJ, Britton KL, Artymiuk PJ, Baker PJ, Sedelnikova SE, Engel PC, Pasquo A, Chiaraluce R, Consalvi V. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure. 1995;3:1147–1158. doi: 10.1016/s0969-2126(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 33.Rice DW, Yip KS, Stillman TJ, Britton KL, Fuentes A, Connerton I, Pasquo A, Scandura R, Engel PC. Insights into the molecular basis of thermal stability from the structure determination of Pyrococcus furiosus glutamate dehydrogenase. FEMS Microbiol Rev. 1996;18:105–117. doi: 10.1111/j.1574-6976.1996.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 34.Harris GW, Pickersgill RW, Connerton I, Debeire P, Touzel JP, Breton C, Perez S. Structural basis of the properties of an industrially relevant thermophilic xylanase. Proteins. 1997;29:77–86. [PubMed] [Google Scholar]
- 35.Wallon G, Kryger G, Lovett ST, Oshima T, Ringe D, Petsko GA. Crystal structures of Escherichia coli and Salmonella typhimurium 3-isopropylmalate dehydrogenase and comparison with their thermophilic counterpart from Thermus thermophilus. J. Mol. Biol. 1997;266:1016–1031. doi: 10.1006/jmbi.1996.0797. [DOI] [PubMed] [Google Scholar]
- 36.Russell RJ, Ferguson JM, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the hyperthermophilic archaeon Pyrococcus furiosus at 1.9 Å resolution. Biochem. 1997;36:9983–9994. doi: 10.1021/bi9705321. [DOI] [PubMed] [Google Scholar]
- 37.Baker D, Sohl JL, Agard DA. A protein folding reaction under kinetic control. Nature. 1992;356:263–265. doi: 10.1038/356263a0. [DOI] [PubMed] [Google Scholar]
- 38.Kelch BA, Agard DA. Mesophile versus thermophile: insights into the structural mechanisms of kinetic stability. J. Mol. Biol. 2007;370:784–795. doi: 10.1016/j.jmb.2007.04.078. [DOI] [PubMed] [Google Scholar]
- 39.Dachs GU, Tupper J, Tozer GM. From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer Drugs. 2005;16(4):349–359. doi: 10.1097/00001813-200504000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Norris JS, Norris KL, Holman DH, El-Zawahry A, Keane TE, Dong JY, Tavassoli M. The present and future for gene and viral therapy of directly accessible prostate and squamous cell cancers of the head and neck. Future Oncol. 2005;1(1):115–123. doi: 10.1517/14796694.1.1.115. [DOI] [PubMed] [Google Scholar]
- 41.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61(7):3022–3026. [PubMed] [Google Scholar]
- 42.Wiedrodt R, Amin K, Kiefer M, Jovanovic VP, Kapoor V, Force S, Chang M, Lanuti M, Black ME, Kaiser LR, Albelda SM. Adenovirus-mediated gene transfer of enhanced Herpes simplex virus thymidine kinase mutants improves prodrug-mediated tumor cell killing. Cancer Gene Ther. 2003;10(5):353–364. doi: 10.1038/sj.cgt.7700589. [DOI] [PubMed] [Google Scholar]
- 43.Rogulski KR, Kim JH, Kim SH, Freytag SO. Glioma cells transduced with an Escherichia coli CD/HSV-1 TK fusion gene exhibit enhanced metabolic suicide and radiosensitivity. Hum. Gene Ther. 1997;8:73–85. doi: 10.1089/hum.1997.8.1-73. [DOI] [PubMed] [Google Scholar]
- 44.Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, Jund R, Mehtali M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000;60:3813–3822. [PubMed] [Google Scholar]
- 45.Adachi Y, Tamiya T, Ichikawa T, Terada K, Ono Y, Matsumoto K, Furuta T, Hamada H, Ohmoto T. Experimental gene therapy for brain tumors using adenovirus-mediated transfer of cytosine deaminase gene and uracil phosphoribosyltransferase gene with 5-fluorocytosine. Hum. Gene Ther. 2000;11:77–89. doi: 10.1089/10430340050016175. [DOI] [PubMed] [Google Scholar]
- 46.Willmon CL, Krabbenhoft E, Black ME. A guanylate kinase/ HSV-1 thymidine kinase fusion protein enhances prodrug-mediated cell killing. Gene Ther. 2006;13:1309–1312. doi: 10.1038/sj.gt.3302794. [DOI] [PubMed] [Google Scholar]
- 47.Kurtz JE, Black ME. Enhancement of suicide gene prodrug activation by random mutagenesis. Methods Mol. Med. 2004;90:331–344. doi: 10.1385/1-59259-429-8:331. [DOI] [PubMed] [Google Scholar]
- 48.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 49.Mahan SD, Ireton GC, Stoddard BL, Black ME. Alanine-scanning mutagenesis reveals a cytosine deaminase mutant with altered substrate preference. Biochem. 2004;43:8957–8964. doi: 10.1021/bi049720z. [DOI] [PubMed] [Google Scholar]
- 50.Kokoris MS, Sabo P, Adman ET, Black ME. Enhancement of tumor ablation by a selected HSV-1 thymidine kinase mutant. Gene Ther. 1999;6:1415–1426. doi: 10.1038/sj.gt.3300966. [DOI] [PubMed] [Google Scholar]
- 51.Dantas G, Kuhlman B, Callender D, Wong M, Baker D. A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J. Mol. Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 53.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 2):484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 54.Brunger AT. Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr D Biol Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 55.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 56.McRee DE. A visual protein crystallographic software system for X11/XView. J. Mol. Graphics. 1992;10:44–46. [Google Scholar]