

Abstract
Krüppel-like factor 4 (KLF4) is an epithelial cell-enriched, zinc finger-containing transcription factor, the expression of which is associated with growth arrest. Previous studies show that constitutive expression of KLF4 inhibits DNA synthesis but the manner by which KLF4 exerts this effect is unclear. In the present study, we developed a system in which expression of KLF4 is controlled by a promoter that is induced upon treatment of cells containing the receptors for the insect hormone, ecdysone, with ponasterone A, an ecdysone analogue. The rate of proliferation of a stably transfected colon cancer cell line, RKO, was significantly decreased following addition of ponasterone A when compared with untreated cells. Flow cytometric analyses indicated that the inducible expression of KLF4 caused a block in the G1/S phase of the cell cycle. A similar block was observed when ecdysone receptor-containing RKO cells were infected with a replication-defective recombinant adenovirus containing an inducible KLF4 and treated with ponasterone A. Results of these studies provide evidence that the inhibitory effect of KLF4 on cell proliferation is mainly exerted at the G1/S boundary of the cell cycle.
Krüppel-like factor 4 (KLF4;1 also called gut-enriched Krüppel-like factor) is a recently identified zinc finger-containing transcription factor that is related to the Drosophila melanogaster segmentation gene product, Krüppel (1–3). It belongs to a rapidly expanding family of mammalian Krüppel-like factors with erythroid Krüppel-like factor (KLF1) (4) serving as a prototype member (5–7). Within this family, KLF4 is most closely related to KLF1 and KLF2 (lung Krüppel-like factor or LKLF) (8). Through studies involving gene knockout, all three KLFs have been shown to be important in regulating development of tissue-specific functions (9–15). KLF4, for example, was shown to be necessary for the development of the barrier function of the skin (15).
Expression of KLF4 is particularly enriched in epithelial tissues such as the intestine and skin (1, 2, 15–17). In the intestinal epithelium, KLF4 is abundantly expressed in the post-mitotic, differentiated epithelial cells outside the proliferating zone of crypt cells (1, 2, 16). In addition, in conjunction with several other KLFs, KLF4 is highly expressed in naive, quiescent B lymphocytes and becomes rapidly down-regulated upon mitogenic activation (18). The in vitro pattern of expression of KLF4 mirrors its in vivo expression. For example, KLF4 is primarily expressed in a growth-arrested state brought on by serum deprivation, contact inhibition, DNA damage, or growth-inhibitory cytokines (1, 19, 20). Conversely, expression of KLF4 is decreased in conditions associated with increased proliferation such as in neoplasm of the intestinal tract (21, 22). These studies, therefore, demonstrate a correlation between KLF4 expression and a process of growth arrest.
Consistent with the notion that KLF4 may be important in regulating growth, previous studies have demonstrated that KLF4 is an inhibitor of cellular proliferation. For example, constitutive expression of KLF4 in transfected fibroblasts or colon cancer cells resulted in the inhibition of DNA synthesis (1, 22). This may in part be explained by the ability of KLF4 to activate the p21WAF1/Cip1 promoter (19), a cell cycle inhibitor, and to suppress the cyclin D1 promoter (23), a positive cell cycle regulator. However, the exact mechanism by which KLF4 inhibits cell proliferation is somewhat unclear, with evidence suggesting that it causes either apoptosis (20) or cell cycle arrest (22). To further define the molecular mechanism by which KLF4 regulates the cell cycle, we developed a system using an inducible promoter to control the expression of KLF4. We show by two independent means that induced KLF4 expression results in an accumulation of cells in the G1 phase of the cell cycle with a concomitant decrease of those in the S phase. The results of our study therefore provide evidence that KLF4 is a cell cycle regulator and does so by creating a block in the G1/S transition phase of the cell cycle.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
pVgRXR, a plasmid engineered to constitutively express both the VgEcR and RXR receptor subunit, was obtained from Invitrogen (Carlsbad, CA). VgEcR was generated by fusing the DNA-binding domain of the D. melanogaster ecdysone receptor (EcR) to a modified transactivation domain of the herpes simplex virus 1 VP16 protein (24, 25). RXR (retinoid X receptor) is the mammalian homologue of USP (ultraspiracle), the natural partner to the Drosophila ecdysone receptor (26, 27). Upon binding to ecdysone, the heterodimeric VgEcR and RXR form a functional ecdysone receptor that is required for the optimal interaction with the ecdysone receptor element (EcRE).
The plasmid pAdLoxEGI-KLF4 was obtained by subcloning the full-length coding region of the KLF4 cDNA into pAdLoxEGI (28). The latter was constructed from the pAdLox plasmid (29) by substituting the ecdysone inducible promoter from pIND (Invitrogen) for the cytomegalovirus promoter and by inserting an expression cassette containing the enhanced green fluorescence protein (EGFP) (30), which was followed by an internal ribosome entry site (31). The KLF4 cDNA was inserted between the XhoI and EcoRI sites in the multiple cloning sites following the internal ribosome entry site.
Transfection and Immunocytochemistry
Transfection was accomplished using the LipofectAMINE reagent from Life Technologies (Gaithersberg, MD). To test the co-expression of EGFP and KLF4, EcR-CHO cells, a Chinese hamster ovary (CHO) cell line containing the stably integrated pVgRXR vector, were cultured on glass coverslips in 6-well dishes. Cells were transfected with pAdLoxEGI-KLF4 and treated for 24 h with 5 μm ponasterone A (Invitrogen). The coverslip was washed twice with phosphate-buffered saline (PBS) and fixed in 3.5% paraformaldehyde in PBS for 8 min followed by washing twice with PBS. Cells were then permeabilized in 0.1% Nonidet P-40 in PBS for 15 min, washed twice with PBS, and incubated in 10% fetal bovine serum (FBS) in PBS for 20 min. Fifty μl of a 1:100 diluted anti-KLF4 serum in PBS containing 10% FBS was added to the coverslip and incubated at 4 °C overnight. The coverslip was washed 3 times with 10% FBS in PBS followed by the addition of 100 μl of a 1:200 dilution of Cy3-labeled secondary antibody and incubated for 1 h at room temperature. The coverslip was then washed with 0.01% Tween 20 in PBS for 30 min and placed face down in 100 μl of mounting media containing 50% glycerol in PBS. Stained cells were visualized under a confocal microscope using filters designed to detect GFP and Cy3. The primary antibody was eliminated in control experiments.
Establishment of Stable Cell Lines
The EcR-RKO/pAdLoxEGI-KLF4 cell line was generated by co-transfecting pVgRXR and pAdLox-EGI-KLF4 at a molar ratio of 1:20 into the RKO human colon cancer cells. Two days following transfection, 150 μg/ml Zeocin was added to the medium to select for resistant clones. To identify clones that contained the inducible KLF4, cells were treated with 5 μm ponasterone A for 24 h. Individual clones that exhibited green fluorescence were expanded and further sorted by Star Plus (Becton Dickinson) following induction to enrich for cells stably expressing EGFP. The level of KLF4 induction was tested by Northern and Western blot analyses upon hormonal treatment.
EcR-RKO cells were similarly obtained as above except that transfection was performed with pVgRXR alone. Zeocin-resistant clones were selected and the degree of RXR expression measured by Western blot analysis.
Generation of Recombinant Adenovirus and Conditions of Infection
The recombinant adenovirus containing EGFP and KLF4 (AdEGI-KLF4) or GFP alone (AdEGI) was generated by Crelox recombination of purified ψ5 viral DNA (29) and pAdLoxEGI-KLF4 or pAdLoxEGI, respectively, as previously described (28, 29). The recombinant products were plaque-purified and expanded by infecting HEK293 cells for 8 h. Forty-eight hours after infection, cells were freeze-thawed 3 cycles to release the viruses, which were purified by CsCl gradient centrifugation to achieve a titer of ~1010 plaque forming units/ml.
EcR-RKO cells were grown to 90% confluence in 10-cm dishes and replenished with fresh media containing 2% FBS followed by the addition of 108 plaque forming units of recombinant virus per dish. Infected cells were incubated at 37 °C for 6–8 h, at which time the media were changed and incubation continued overnight. Cells were then treated with 5 μm ponasterone A for 24 h and then collected for further analyses.
Northern Blot and Western Blot Analyses
RNA was extracted from cells using the TriZol reagent (Life Technologies) following the manufacturers recommendations. Twenty μg of total RNA was resolved by electrophoresis in 1.2% agarose gels containing 2.4 m formaldehyde and transblotted onto nylon membranes (Hybond-N; Amersham Pharmacia Biotech). Hybridization and washing were performed under high stringency conditions using radioactively labeled KLF4 and glyceraldehyde-3-phosphate dehydrogenase cDNA probes.
Western blot analyses were performed using standard procedures. Protein samples were dissolved in a loading buffer (60 mm Tris-HCl, pH 6.8, 2% SDS, 100 mm dithiothreitol, and 0.01% bromphenol blue), heated at 100 °C for 3 min, and loaded onto a SDS-polyacrylamide gel in running buffer containing 25 mm Tris-HCl, pH 8.3, 250 mm glycine, and 0.1% SDS. At the completion of electrophoresis, proteins were electrophoretically transferred to a membrane, which was immunoblotted with a rabbit anti-KLF4 serum (1:1,000 dilution; Ref. 1) or anti-RXR serum (1:1,000 dilution; Santa Cruz). Following incubation with the secondary antibody (horseradish peroxidase-conjugated donkey anti-rabbit IgG; Santa Cruz), KLF4 or RXR was visualized with enhanced chemiluminescence (Amersham Pharmacia Biotech).
Cell Proliferation Assays
EcR-RKO/pAdLoxEGI-KLF4 cells were plated on 60-mm dishes at a density of 2.8 × 105 cells/dish. After 24 h, ponasterone A was added to a final concentration of 5 μm. Media containing ponasterone A were changed every other day. Control cells received vehicle (ethanol) only. Cell numbers were measured with a hemocytometer daily up to 8 days following seeding.
MTS assays were performed based using the manufacturers recommendations (Promega, Madison, WI). EcR-RKO and EcR-RKO/pAdLox-EGI-KLF4 cells were plated in 96-well microtiter plates at a density of 1.0 × 103 cells/well in Dulbecco’s modified Eagle’s medium with 10% FBS and 150 μg/ml Zeocin. After 24 h, the medium was changed to Dulbecco’s modified Eagle’s medium containing 10% FBS, 150 μg/ml zeocin, and either 5 μm ponasterone A or vehicle alone. The number of cells at this time (t = day 0) and at 1, 3, and 5 days was determined using the colorimetric MTS assay. Results are depicted as absorbance at 490 nm as a function of time.
Cell Cycle Analysis
DNA content was measured following staining of cells with propidium iodide. EcR-RKO/pAdLoxEGI-KLF4 cells were treated with 5 μm ponasterone A or vehicle alone for 24 h. Cells were subsequently trypsinized, washed once in cold PBS, and fixed in 70% ethanol at −20 °C overnight. Fixed cells were pelleted and stained in the propidium iodide solution (50 μg/ml propidium iodide, 50 μg/ml RNase A, 0.1% Triton X-100, and 0.1 mm EDTA) in the dark at 4 °C for 1 h prior to flow cytometric quantification of DNA by a FACScan (Becton Dickinson). Similarly, FACS analyses were conducted in EcR-RKO cells infected with recombinant AdLoxEGI-KLF4 or AdLoxEGI viruses followed by treatment with 5 μm ponasterone A or vehicle alone for 24 h.
RESULTS
To investigate the mechanism by which KLF4 regulates the cell cycle, we first established an inducible system in which KLF4 expression could be controlled. We generated a DNA construct called pAdLoxEGI-KLF4 that would produce a bicistronic message of enhanced green fluorescence protein (EGFP) and KLF4 separated by an internal ribosomal entry site (31). Expression of EGFP and KLF4 was controlled by a promoter that contained a response element to a hormone-activated EcR (24). A CHO cell line, called EcR-CHO, that had a stably integrated dimeric ecdysone receptor and RXR (26, 27) derived from the plasmid pVgRXR was transiently transfected with pAdLoxEGI-KLF4 and treated with an ecdysone analogue, ponasterone A. As seen in Fig. 1, GFP and KLF4 were produced in the same transfected cells (panels A and B, respectively), as evidenced by direct fluorescence visualization of the former and by immunocytochemical staining of the latter. As a control, transfected cells (Fig. 1C) from which the primary KLF4 antibody was eliminated during staining lacked any fluorescence (Fig. 1D). These results indicate that the pAdLoxEGI-KLF4 construct was able to produce both proteins in transfected cells upon induction.
Fig. 1. Inducible expression of EGFP and KLF4 in transiently transfected cells.
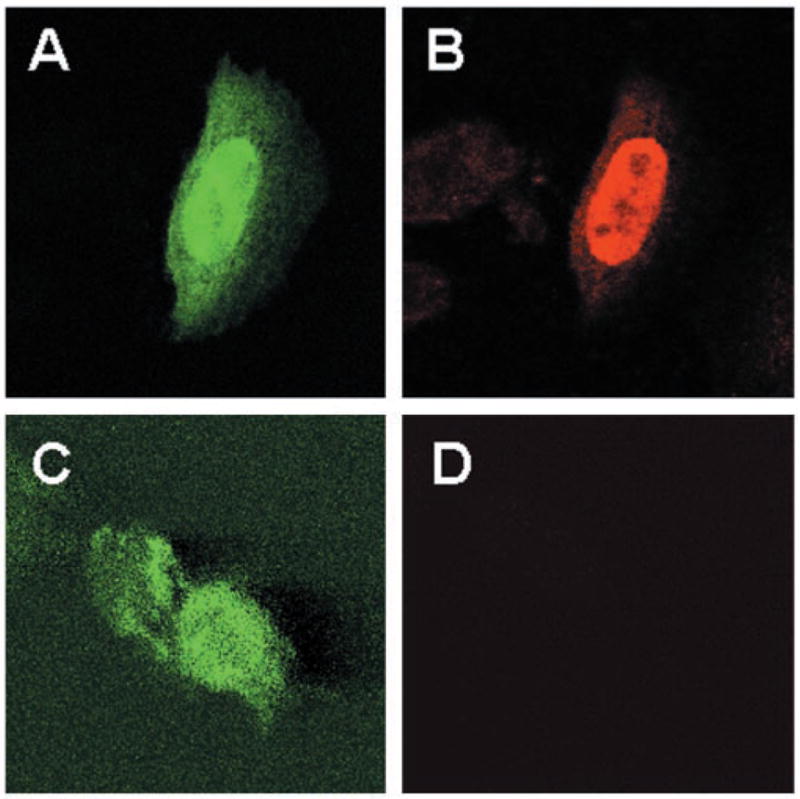
EcR-CHO cells were transfected with pAdLoxEGI-KLF4 and treated with 5 μm ponasterone A for 24 h. Cells were fixed and stained with an anti-KLF4 antibody (panel B) or without any primary antibody (panel D), followed by a Cy3-labeled secondary antibody. Shown in panels A and B is a representative cell that contained EGFP (green) and KLF4 (red), respectively. In contrast, neither of the two green cells in panel C stained red in the absence of the primary antibody (panel D) (magnification, ×100).
We then established a cell line that contained stably integrated pVgRXR and pAdLoxEGI-KLF4 by co-transfecting a human colon cancer cell line, RKO. Clonal derivatives were selected by the antibiotic Zeocin. We chose RKO cells because of their exceedingly low level of KLF4 mRNA, which is probably due to mutations in upstream genes that regulate KLF4 expression (32). A stably transfected clone, called EcR-RKO/pAd-LoxEGI-KLF4, was treated with ponasterone A or vehicle alone for 24 h and visualized with a fluorescence microscope. As shown in Fig. 2A, the addition of ponasterone A resulted in the appearance of numerous green cells in the culture (+PA), whereas, the uninduced control cells exhibited no fluorescence (−PA). The addition of ponasterone A also resulted in a significant increase in the level of the KLF4 transcripts as demonstrated by Northern blot analysis (Fig. 2B).
Fig. 2. Inducible expression of KLF4 in stably transfected RKO cells.
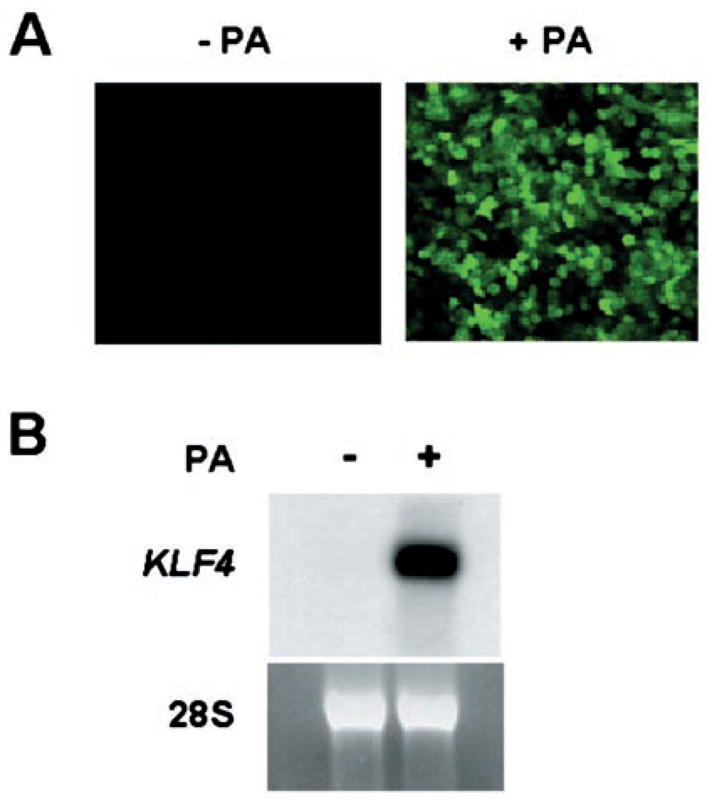
A stable RKO cell line (EcR-RKO/pAdLoxEGI-KLF4) was established by co-transfection with pVgRXR and pAdLoxEGI-KLF4. After treatment with or without 5 μm ponasterone A (PA) for 24 h, cells were inspected under an inverted fluorescence microscope for the presence of GFP (panel A). Panel B shows the result of a Northern blot analysis of RNA isolated from uninduced (−) and induced (+) cells, and probed with a labeled KLF4 cDNA fragment (upper panel). A photograph of the 28 S ribosomal RNA (lower panel) from both sets of cells is included to indicate equal loading.
To begin investigating the effect of induced KLF4 expression on cell proliferation, we seeded EcR-RKO/pAdLoxEGI-KLF4 cells in low densities and treated them with ponasterone A or vehicle alone for up to 8 days. Cell numbers were assessed by direct daily counting (Fig. 3A) or by MTS assay every other day (Fig. 3B). As seen, the ponasterone A-treated cells did not proliferate during the first 4 days of treatment when compared with the control cells. After 4 days of treatment, the number of induced cells did begin to increase but at a slower rate compared with the uninduced cells. As demonstrated by the Western blot analysis in Fig. 3C, the level of KLF4 protein was low in uniduced cells (day 0) but was increased 1 day following induction and maintained at a similar level up to 6 days. These results suggest that the decreased proliferation in induced cells is a consequence of KLF4 production.
Fig. 3. The effect of induced KLF4 expression on proliferation of RKO cells.
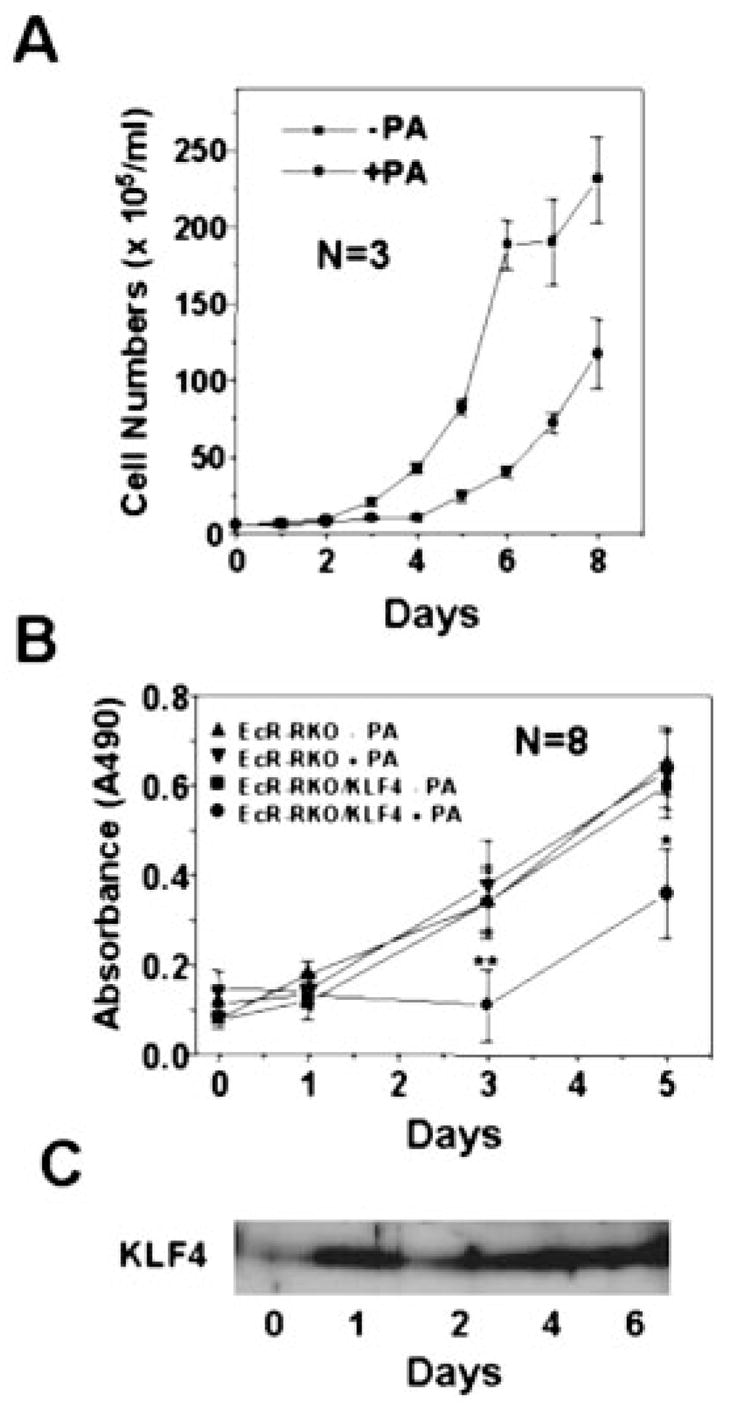
In panel A, 2.8 × 105 EcR-RKO/pAdLoxEGI-KLF4 cells were seeded and continuously cultured in 60-mm dishes with or without 5 μm ponasterone A for up to 8 days. The media were changed every other day. Cells were counted with a hemocytometer daily. Cell numbers are expressed as mean ± S.D. (bars). In panel B, EcR-RKO and EcR-RKO/pAdLoxEGI-KLF4 (labeled as EcR-RKO/KLF4 in the figure) cells were plated in 96-well microtiter plates at a density of 1 × 103 cells per well. After 24 h, fresh media containing either 5 μm ponasterone A or vehicle only were added. The number of cells was determined using the colorimetric MTS assay on days 0, 1, 3, and 5. Results are depicted as mean ± S.D of absorbance at 490 nm as a function of time. *, p < 0.01; **, p < 0.0001 by paired Student t test. Panel C is a Western blot analysis of proteins extracted from induced cells for the presence of KLF on the days indicated.
To address the mechanism by which KLF4 inhibits cell proliferation, we performed FACS analyses of EcR-RKO/pAdLox-EGI-KLF4 cells treated or not with ponasterone A for 24 or 48 h. As shown in Fig. 4A, a great majority of cells exhibited green fluorescent after 24 h of ponasterone A treatment. This was accompanied by a change in the cell cycle profile as illustrated in Fig. 4B. Specifically, the induction of KLF4 caused a statistically significant increase in the percentage of cells in the G1 phase of the cell cycle and a statistically significant decrease in the percentage of cells in the S phase (Fig. 4C). The difference in the percentage of cells in the G2 phase between treated and control groups was not significant. In addition, FACS analyses showed no evidence of apoptosis in the induced cells, which would have been manifested by a DNA content, that is less than G1 as in Fig. 4B. The results obtained after 48 h of induction were similar to those at 24 h (results not shown). Importantly, accompanying the G1/S block associated with hormonal induction was a concomitant increase in the amount of p21WAF1/Cip1 mRNA (Fig. 4C, inset). These results suggest that the ability of KLF4 in blocking the cell cycle is mediated by p21WAF1/Cip1.
Fig. 4. The effect of induced KLF4 expression on the cell cycle.
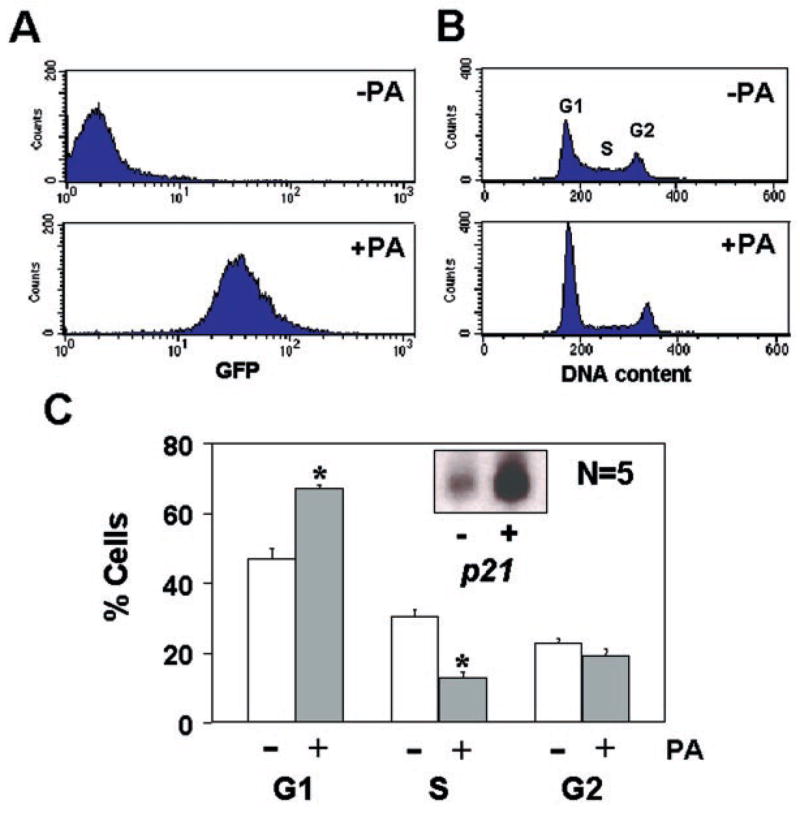
EcR-RKO/pAdLoxEGI-KLF4 cells were cultured to ~50% confluency and treated with or without 5 μm ponasterone A for 24 h. Cells were fixed and stained with propidium iodide, and then analyzed by FACS. Panel A shows the result of FACS based on the intensity of green fluorescence in uninduced (−PA) and induced (+PA) cells. Panel B shows the DNA content as revealed by propidium iodide staining in both conditions. DNA contents corresponding to the 3 phases (G1, S, and G2) of the cell cycle are labeled as such. In panel C, the mean percentages of cells with DNA content in each of the 3 phases of the cell cycle under the uninduced or induced condition over five independent determinations were shown as mean ± S.D. (bars). *, p < 0.001. Inset shows the result of a Northern blot analysis for p21WAF1/Cip1 in the absence (−) or presence (+) of ponasterone A.
As an alternative and independent means to measure the effect of KLF4 on the cell cycle, we constructed a recombinant adenovirus, called AdEGI-KLF4, using the pAdLoxEGI-KLF4 plasmid as a shuttle vector (see “Experimental Procedures”). We also established a RKO cell line, called EcR-RKO, which had a stably integrated pVgRXR construct (Fig. 5A, lane 3). Following infection with the AdEGI-KLF4 recombinant virus, EcR-RKO cells were treated with ponasterone A or vehicle alone for 24 h and the degree of KLF4 expression assessed by Northern and Western blot analyses. As shown in Fig. 5B, both the levels of KLF4 mRNA and protein were significantly increased in infected and induced cells when compared with infected but uninduced cells (Fig. 5B, upper and lower panel, respectively). The induced KLF4 expression was accompanied by the appearance of green fluorescence in cells treated with ponasterone A (Fig. 5C). To control for a possible effect of EGFP on the cell cycle, we also generated a recombinant adenovirus, called AdEGI that contained only EGFP. As seen in Fig. 5D, EcR-RKO cells infected with the AdEGI virus and induced with ponasterone A also exhibited intense green fluorescence at 24 h. Importantly, FACS analysis of infected cells showed only those infected by AdEGI-KLF4 and induced with ponasterone A had a statistically significant increase in the G1 population and a statistically significant decrease in the S population when compared with the infected but uninduced cells (Fig. 5E). There was no difference in the cell cycle profiles of AdEGI virus-infected cells between induced and uninduced conditions (Fig. 5F). These results suggest that the alteration in cell cycle profile upon ponasterone A addition is the result of induction in KLF4 and not in EGFP expression.
Fig. 5. The effect of inducible expression of KLF4 in cells infected with recombinant adenoviruses containing EGFP and KLF4 or EGFP alone.
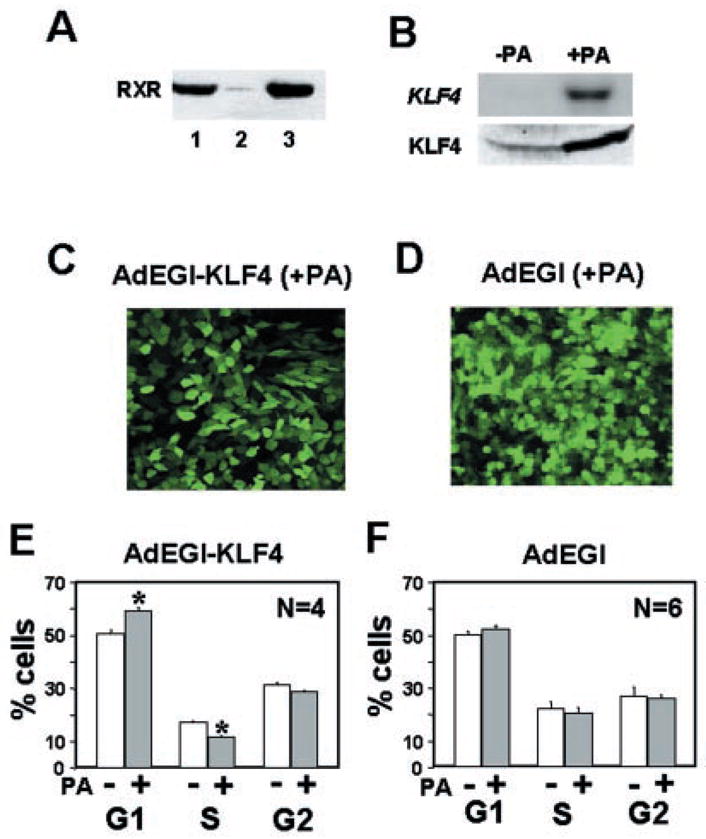
Panel A is a Western blot analysis for RXR production in CHO cells with stably transfected pVgRXR (lane 1), untransfected RKO cells (lane 2), and the stably transfected RKO cells, EcR-RKO (lane 3). In panel B, EcR-RKO cells were infected with the recombinant AdEGI-KLF4 virus and treated (+PA) or not (−PA) with ponasterone A for 24 h and then analyzed for the RNA (upper panel) and protein (lower panel) content for KLF4 by Northern and Western blotting, respectively. In panels C and D, EcR-RKO cells were infected with AdEGI-KLF4 and AdEGI, respectively, and treated with 5 μm ponasterone A for 24 h. Shown are cells observed under a fluorescence microscope. In panels E and F, EcR-RKO cells were infected with AdEGI-KLF4 and AdEGI, respectively, and treated or not with 5 μm ponasterone A for 24 h. Cells were stained with propidium iodide and the DNA content analyzed by FACS. Shown are the mean and S.D. of percentages of cells in the 3 phases of the cell cycle. *, p < 0.001.
DISCUSSION
KLF4 was first identified some 5 years ago as a gene whose expression is temporally associated with growth arrest (1). This correlation has been documented in several independent in vitro conditions (1, 19, 20, 23). In vivo, the predominant expression of KLF4 in differentiated cells of several epithelial tissues also suggests that it is a growth arrest-specific gene (1, 2, 15, 16, 33). It is particularly telling that KLF4, along with several other Krüppel-like factor-encoding genes, is highly expressed in naive, quiescent B lymphocytes and is rapidly down-regulated upon activation due to mitogenic stimulation (18). These observations suggest that KLF4 may have an inhibitory function in regulating cell proliferation. Consistent with this notion, KLF4 was shown to inhibit DNA synthesis when constitutively produced in transfected cells (1).
The present study established the mechanism by which KLF4 exerts its negative effect on cell growth. This was accomplished by the development of an inducible system for KLF4 expression. By either stable transfection or adenovirus-mediated infection of RKO cells, we demonstrated that induced expression of KLF4 caused a significantly decreased rate of proliferation, which was likely due to a block in the cell cycle at the G1/S boundary. The result of our study is consistent with that from a previous one, which examined the effect of constitutive KLF4 expression in a stably transfected colon cancer cell line, HT-29 (22). It is also consistent with the finding that, among the genes regulated by KLF4 are p21WAF1/Cip1 and cyclin D1, two genes with key, albeit opposite, effects in controlling G1/S transition in the cell cycle (35–37). As KLF4 is an activator of transcription of p21WAF1/Cip1 (19) and a suppressor of transcription of cyclin D1 (23), a logical consequence of KLF4s activation is the impediment in the transition from G1 to S phase as observed in the present study. The physiological implication of our finding extends our previous observation that cell cycle arrest due to DNA damage is accompanied by p53-dependent activation of KLF4 expression (19). Moreover, an activated KLF4 transcriptionally induces p21WAF1/Cip1 expression. This is further supported by the present study showing that the level of p21WAF1/Cip1 transcript is significantly increased due to the induction of KLF4 (Fig. 4C). Combining the results of these studies, it appears likely that KLF4 is an essential member of the p53 tumor suppressor network of cell cycle regulators.
A recent study showed that expression of KLF4 is up-regulated by treatment of the human colon cancer cell line, HT-29, with the cytokine interferon-γ (20). In this system, interferon-γ induced growth arrest in HT-29 cells by causing apoptosis. Moreover, transient transfection of several colon cancer cell lines with a KLF4-expressing plasmid was shown to promote apoptosis as evidenced by DNA fragmentation (20). It was proposed that KLF4 may be a downstream target gene of interferon-γ and may mediate the apoptotic effect of interferon-γ. In our system, however, we did not observe any evidence of apoptosis by FACS or DNA staining (results not shown) during the first 48 h of induction in either stably transfected or infected cells. As the same group has reported in a different study that stable expression of KLF4 in HT-29 cells caused cell cycle arrest but not apoptosis (22), it is unclear at this time whether KLF4 is in fact a pro-apoptotic factor.
The negative effect of KLF4 on the cell cycle progression suggests that KLF4 may play an important role in regulating the transition of cells from a proliferative to a non-proliferative state such as those observed in vitro and in vivo (1, 2, 15, 16, 18–20, 23, 33). It may also be reasonable to speculate that one of KLF4s functions in terminally differentiated epithelial cells in organs such as the gut or skin is to prevent the cells from re-entering the cell cycle. Conversely, in situations where there is increased proliferation such as that encountered in neoplasm, one may expect to see a decrease in KLF4 expression, which may in turn contribute to the tumorigenic phenotype. Indeed, expression of KLF4 is diminished in benign intestinal adenomas when compared with the surrounding normal intestinal mucosa in both mouse and human models of a hereditary polyposis syndrome (21). Similarly, levels of KLF4 mRNA have been shown to be lower in sporadic cases of colonic adenomas and carcinomas (22). However, it should be cautioned that the expression pattern of KLF4 in cancer tissues might not always be consistent with its putative role as a tumor suppressor. For example, increased expression of KLF4 has been observed in dysplastic squamous epithelium and squamous cell carcinoma of the oral cavity (38), and in breast cancer (39). Moreover, evidence suggests that KLF4 may be deregulated or misexpressed in certain tumorous tissues (22, 38). A potential explanation for the apparent discrepancies stated above is that certain tumors may contain mutated forms of KLF4, which may exert a dominant negative effect on cell function. Previous studies indicating that KLF4 interacts with multiple regulatory proteins including Sp1 (40), p53 (19), Zf9 (41), and p300/CBP (34) distinctly suggest that this latter hypothesis may be a possibility.
Footnotes
This work was supported in part by National Institutes of Health Grants DK52230 and CA84197 (to V. W. Y.).
The abbreviations used are: KLF4, Krüppel-like factor 4; AdEGI, adenovirus containing EGFP; AdEGI-KLF4, adenovirus containing EGFP and KLF4; CHO, Chinese hamster ovary; EcR, ecdysone receptor; EGFP, enhanced green fluorescence protein; FACS, fluorescence activated cell sorting; FBS, fetal bovine serum; LKLF, lung Krüppel-like factor; PA, ponasterone A; PBS, phosphate-buffered saline; RXR, retinoid X receptor; EcRE, ecdysone receptor element; MTS, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
References
- 1.Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 3.Schuh R, Aicher W, Gual U, Cote S, Preiss A, Maier D, Seifert E, Nauber U, Shroder C, Kemler R, Jackle H. Cell. 1986;47:1025–1032. doi: 10.1016/0092-8674(86)90817-2. [DOI] [PubMed] [Google Scholar]
- 4.Miller IJ, Bieker JJ. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner J, Crossley M. Trends Biochem Sci. 1999;24:236–240. doi: 10.1016/s0968-0004(99)01406-1. [DOI] [PubMed] [Google Scholar]
- 6.Philipsen S, Suske G. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang DT, Pevsner J, Yang VW. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KP, Kern CB, Crable SC, Lingrel JB. Mol Cell Biol. 1995;15:5957–5965. doi: 10.1128/mcb.15.11.5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 10.Perkins AC, Sharpe AH, Orkin SH. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 11.Kuo CT, Veselits ML, Leiden JM. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 12.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wani MA, Means RT, Jr, Lingrel JB. Transgenic Res. 1998;7:229–238. doi: 10.1023/a:1008809809843. [DOI] [PubMed] [Google Scholar]
- 14.Wani MA, Wert SE, Lingrel JB. J Biol Chem. 1999;274:21180–21185. doi: 10.1074/jbc.274.30.21180. [DOI] [PubMed] [Google Scholar]
- 15.Segre JA, Bauer C, Fuchs E. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 16.Ohnishi S, Laub F, Matsumoto N, Asaka M, Ramirez F, Yoshida T, Terada M. Dev Dyn. 2000;217:421–429. doi: 10.1002/(SICI)1097-0177(200004)217:4<421::AID-DVDY9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 17.Jenkins TD, Opitz OG, Okano J, Rustgi AK. J Biol Chem. 1998;273:10747–10754. doi: 10.1074/jbc.273.17.10747. [DOI] [PubMed] [Google Scholar]
- 18.Glynne R, Ghandour G, Rayner J, Mack DH, Goodnow CC. Immunol Rev. 2000;176:216–246. doi: 10.1034/j.1600-065x.2000.00614.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen ZY, Shie J, Tseng C. FEBS Lett. 2000;477:67–72. doi: 10.1016/s0014-5793(00)01764-6. [DOI] [PubMed] [Google Scholar]
- 21.Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. FEBS Lett. 2000;476:203–207. doi: 10.1016/s0014-5793(00)01727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Am J Physiol. 2000;279:G806–814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- 23.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Nucleic Acids Res. 2000;28:2969–2976. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.No D, Yao TP, Evans RM. Proc Natl Acad Sci U S A. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cress WD, Triezenberg SJ. Science. 1991;251:87–90. doi: 10.1126/science.1846049. [DOI] [PubMed] [Google Scholar]
- 26.Yao TP, Forman BM, Jiang Z, Cherbas L, Chen JD, McKeown M, Cherbas P, Evans RM. Nature. 1993;366:476–479. doi: 10.1038/366476a0. [DOI] [PubMed] [Google Scholar]
- 27.Yao TP, Segraves WA, Oro AE, McKeown M, Evans RM. Cell. 1992;71:63–72. doi: 10.1016/0092-8674(92)90266-f. [DOI] [PubMed] [Google Scholar]
- 28.Johns DC, Marx R, Mains RE, O’Rourke B, Marban E. J Neurosci. 1999;19:1691–1697. doi: 10.1523/JNEUROSCI.19-05-01691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang G, Gurtu V, Kain SR. Biochem Biophys Res Commun. 1996;227:707–711. doi: 10.1006/bbrc.1996.1573. [DOI] [PubMed] [Google Scholar]
- 31.Johns DC, Nuss HB, Marban E. J Biol Chem. 1997;272:31598–31603. doi: 10.1074/jbc.272.50.31598. [DOI] [PubMed] [Google Scholar]
- 32.Dang DT, Mahatan CS, Dang LH, Agboola IA, Yang VW. Oncogene. 2001 doi: 10.1038/sj.onc.1204645. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panigada M, Porcellini S, Sutti F, Doneda L, Pozzoli O, Consalez GG, Guttinger M, Grassi F. Mech Dev. 1999;81:103–113. doi: 10.1016/s0925-4773(98)00237-8. [DOI] [PubMed] [Google Scholar]
- 34.Geiman DE, Ton-That H, Johnson JM, Yang VW. Nucleic Acids Res. 2000;28:1106–1113. doi: 10.1093/nar/28.5.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donjerkovic D, Scott DW. Cell Res. 2000;10:1–16. doi: 10.1038/sj.cr.7290031. [DOI] [PubMed] [Google Scholar]
- 36.Harper JW. Cancer Surv. 1997;29:91–107. [PubMed] [Google Scholar]
- 37.Sherr CJ. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 38.Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. Cell Growth & Differ. 1999;10:423–434. [PubMed] [Google Scholar]
- 39.Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, Ruppert JM. Cancer Res. 2000;60:6488–6495. [PubMed] [Google Scholar]
- 40.Zhang W, Shields JM, Sogawa K, Fujii-Kuriyama Y, Yang VW. J Biol Chem. 1998;273:17917–17925. doi: 10.1074/jbc.273.28.17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okano J, Opitz OG, Nakagawa H, Jenkins TD, Friedman SL, Rustgi AK. FEBS Lett. 2000;473:95–100. doi: 10.1016/s0014-5793(00)01468-x. [DOI] [PubMed] [Google Scholar]