

Abstract
We examined the expression of GKLF (gut-enriched Krüppel-like factor), a recently identified zinc finger-containing transcription factor, in mice during development using the ribonuclease protection assay. In the adult, the level of GKLF transcript is abundant throughout the gastrointestinal tract. Between embryonic days 10 and 19 (E10 and E19) of development, the initial level of whole embryo GKLF transcript is low but begins to rise on E13 and peaks on E17. In the newborn, GKLF transcript level is higher in the colon than in the small intestine although the levels in both organs rise with increasing age. Expression of GKLF was also examined in the intestinal tract of the Min mouse, a model of intestinal tumorigenesis. The level of GKLF transcript is significantly decreased in the intestine of Min mice during a period of tumor formation when compared to age-matched control littermates. Our findings indicate that GKLF expression correlates with certain periods of gut development and is down-regulated during intestinal tumorigenesis, suggesting that GKLF may play a role in gut development and/or tumor formation.
Keywords: Gut-enriched, Krüppel-like factor, Gastrointestinal tract, Development, Tumorigenesis, Min mouse, Ribonuclease protection assay
1. Introduction
The mammalian intestinal epithelium is divided into several distinct compartments within which fundamentally different cellular processes are observed [1,2]. Stem cells reside in the intestinal crypts where they rapidly proliferate to produce descendants that migrate along the crypt-to-villus axis. During migration, cells cease to divide and differentiate into mature cells with characteristics of enterocytes. At the apex of the villus, cells undergo programmed cell death and are exfoliated. This process of proliferation, migration, differentiation, and exfoliation is completed in 3–5 days in the small intestine of the adult mouse [3]. The intestinal epithelium therefore represents an attractive system in which the dynamics of these important biological processes can be studied.
The complex structure of the adult intestinal epithelium is established during late fetal development. In the mouse, the gut endoderm is poorly differentiated before embryonic day 15 (E15). Between E15 and E18, the pseudostratified gut epithelium undergoes dramatic remodeling which is coupled with cytodifferentiation to form a simple columnar epithelium [4]. Accompanying this process is the induction of intestine-specific genes that encode proteins necessary for intestinal function following birth [5]. Development of the adult crypt structure occurs during the first two weeks after birth [6]. Thereafter, dramatic changes in gene expression are seen during the suckling-weaning transition period and after which the adult phenotype is eventually established [7]. It is now widely accepted that gene transcription plays an essential role in the development of gut epithelial function and many recent studies have begun to unravel the mechanisms controlling gut-specific gene expression [5].
Because of its rapid turnover and exposure to environmental carcinogens, the gut epithelium is relatively susceptible to neoplastic transformation. This is exemplified by the high prevalence of colon cancer in humans, especially in the western world [8]. Recently, the genetic basis of colon cancer has partially been deciphered by the identification of gene mutations that occur in families with a hereditary predisposition to colon cancer. A well-known example is the autosomal dominant familial adenomatous polyposis (FAP) syndrome [9]. Individuals affected by FAP carry a single mutant allele of the adenomatous polyposis coli (APC) gene [10,11]. Importantly, a murine model of FAP was independently developed in C57BL/6J mice that had been treated with the mutagen ethy-nitrosourea and bred for germline transmission [12]. The mutation that predisposes the formation of intestinal polyps in this Min (multiple intestinal neoplasia) mouse model was identified as the murine homologue of human APC [13]. Thus, an analysis of this mouse model should greatly facilitate our understanding of the molecular mechanisms responsible for intestinal neoplasia.
We recently identified and isolated a murine cDNA clone, named GKLF (gut-enriched Krüppel-like factor), that encodes a novel transcription factor with three C2H2 zinc fingers of the Krüppel type [14]. Expression of GKLF is enriched in the intestinal tract with the highest level of transcript found in the differentiated epithelial cells of the colonic mucosa. In cell culture, expression of GKLF is induced under conditions that promote growth arrest such as serum deprivation and contact inhibition. Moreover, enforced expression of GKLF in transfected cells results in the inhibition of DNA synthesis. Taken together, these observations suggest that GKLFs have an important function in regulating growth of intestinal epithelial cells. Subsequently, GKLF was independently identified by a second group and shown to be expressed also at high levels in the epidermal layers of the skin [15]. These results suggest that GKLF may also be involved in the terminal differentiation of select epithelial tissues.
To further understand the function of GKLF, we examined its expression during development and intestinal tumorigenesis. We show that changes in GKLF expression correlate with an important landmark period of intestinal development – during morphogenesis of the embryonic gut epithelium. Moreover, we show that expression of GKLF is significantly decreased in the intestine of the Min mouse during a period in which tumor formation occurs. These findings are suggestive of a purported function of GKLF in regulating growth and differentiation of intestinal epithelial cells.
2. Materials and methods
2.1. Animals and preparation of RNA from tissues
Wild-type (+/+) and Min (Min/+) C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The Min pedigree was maintained by crossing Min/+ males with C57BL/6J +/+ females. The Min/+ animals were identified by an allele-specific polymerase chain reaction as previously described [16]. +/+ littermates were used as controls. All animals were killed by CO2 asphyxiation in accordance with the protocol of the Johns Hopkins University. Total RNA was extracted from snap-frozen whole embryos or various segments of the intestine using the TRIzol Reagent purchased from LIFE Technologies (Gaithersburg, MD). After determining the concentration of the RNA samples by optical density measurement, their integrity and quantity were verified with denaturing agarose gel electrophoresis before the ribonuclease protection assay.
2.2. Ribonuclease protection assay (RPA)
An ApaI-BspMI restriction endonuclease fragment of the GKLF cDNA between nt 1360 and 1570 [14] was subcloned into the plasmid pBluescript (Stratagene (La Jolla, CA)) and used to generate a 32P-labeled antisense RNA probe using T3 RNA polymerase. The pTRI-Actin plasmid- (Ambion Inc. (Austin, TX)) containing part of the mouse β-actin cDNA was used to generate an antisense β-actin probe with T3 RNA polymerase and used as a control for RNA loading in all experiments. Probes were synthesized from linearized plasmid templates by in vitro transcription with the designated RNA polymerase and [α-32P]UTP (NEN-DuPont (Boston, MA)) using the MAXIscript protocol provided by Ambion Inc. The size of the synthesized probe for GKLF and β-actin was 277 nt and 300 nt, respectively. The size of the resultant fragment following ribonuclease protection assay was 220 nt and 250 nt for GKLF and β-actin, respectively.
RPA was performed according to the protocol provided by Ambion Inc. Briefly, 20 μg of total RNA were mixed with 3 × 105 cpm of labeled probe (GKLF or β-actin) and precipitated with ethanol after addition of NH4OAc to 0.5 M. A control reaction containing 20 μg of yeast tRNA instead of mouse RNA was performed in all experiments. The precipitated samples were collected by centrifugation and suspended in 20 μl of hybridization buffer containing 300 mM NaOAc, 100 mM NaCitrate, pH 6.4, 1 mM EDTA, and 80% formamide. Following heating to 95°C for 5 min to denature the RNA, samples were annealed at 55°C for 24 h. At the completion of hybridization, 200 μl of a diluted RNase mixture containing a 1:50 dilution of a mixture of 0.5 mg/ml RNase A and 10000 units/ml RNase T1 were added to each sample and incubated at 37°C for 30 min. The reactions were stopped by the addition of 300 μl of an RNase inactivation/precipitation solution (Ambion Inc.) and the samples precipitated with ethanol. The protected products were resolved on a 6% sequencing gel and visualized by autoradiography. Radioactively labeled RNA size markers provided by the manufacturer were also run on the same gel.
3. Results
Ribonuclease protection assay (RPA) was first performed on total RNA isolated from various regions of the gastrointestinal tract of a 4 month-old wild-type C57BL/6J mouse using a 32P-labeled riboprobe prepared from GKLF or β-actin cDNA. Lane 8 of Fig. 1 shows the size of the probe – 277 nt for GKLF (upper panel) and 300 nt for β-actin (lower panel). When various segments of intestinal tissues were analyzed, a single, prominent protected band of 220 nt was noted for GKLF (arrow, upper panel). This size is consistent with the predicted size based on the cDNA sequence. Similarly, a protected fragment of 250 nt was noted when samples were hybridized to the β-actin probe (arrow, lower panel). No protected fragments were seen when yeast tRNA was hybridized to either the GKLF or β-actin probe (lane 7). Fig. 1 shows that the level of GKLF transcript is high throughout the gastrointestinal tract in the 4 month-old mouse with the exception of the proximal small intestine, which contains a slightly lower amount of GKLF mRNA. In addition, the liver contains no GKLF transcript, an observation consistent with our previous finding [14]. Overall, the relative amounts of GKLF transcript in the intestinal tissues as revealed by RPA are in accord with the results of our previous study using Northern blot analysis [14].
Fig. 1.
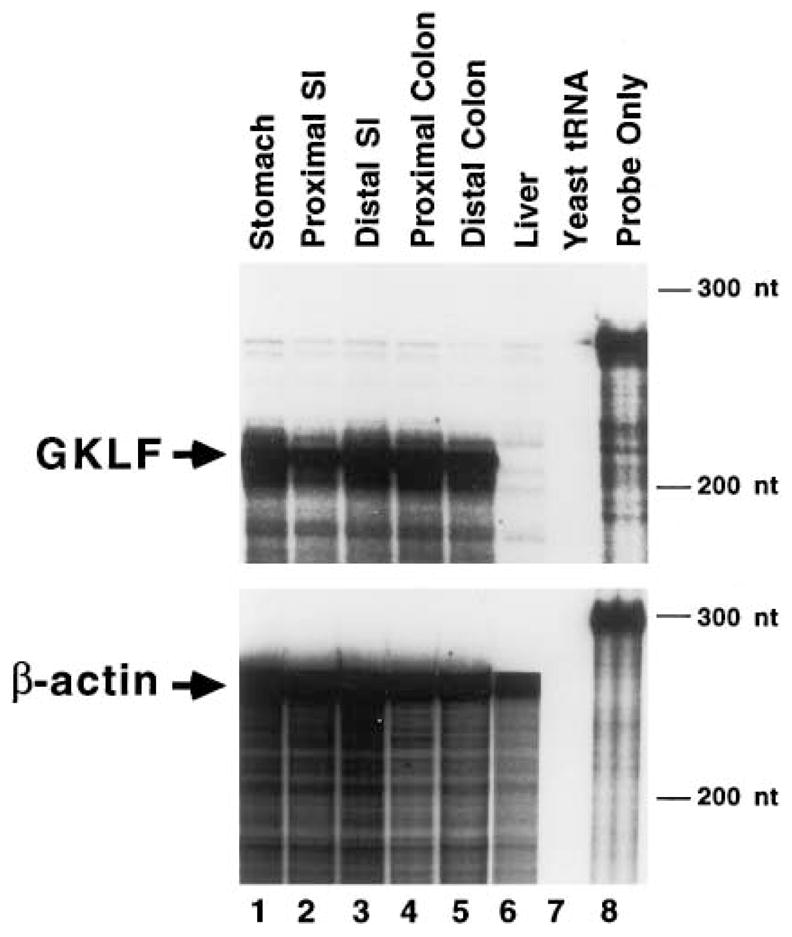
RPA of GKLF transcripts in the adult mouse gastrointestinal tract. Tissues were obtained from a 4 month-old wild-type C57BL/6J mouse. SI is small intestine. Twenty μg of total RNA from each tissue were hybridized to GKLF or β-actin riboprobe. Arrows indicate the protected fragments. Sizes of molecular mass markers are shown on the right. Lane 7 is a control containing yeast tRNA only and lane 8 is probe only.
The expression of GKLF in whole mouse embryos during development was subsequently determined between E10 and E19. RNA was isolated from pooled embryos during each day of the development and multiple pools were independently analyzed. Shown in Fig. 2 is a representative result of these studies. The result indicates that GKLF transcript is very low from E10 to E12, and only begins to rise on E13. The level of the transcript peaks on E17 before decreasing moderately by E19. The absolute level of GKLF transcript at E17 is perhaps even higher than shown in Fig. 2, considering that RNA from this day is underloaded (lane 13, lower panel). As a reference, RNA from various regions of a 4 week-old mouse was also analyzed in the same experiment (lanes 2 to 5).
Fig. 2.
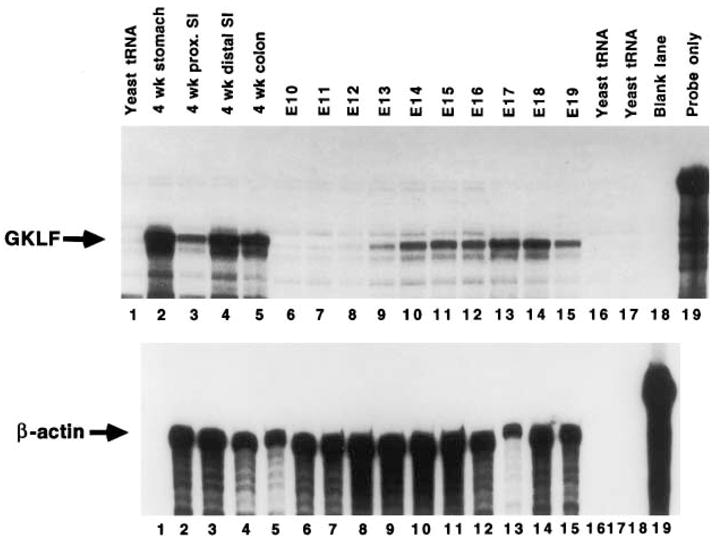
RPA of GKLF transcripts during fetal development. Total RNA was extracted from whole mouse embryos between E10 and E19 of development. Twenty μg from each day were used in the experiment to measure either GKLF or β-actin transcripts. As a comparison, 20 μg of RNA from tissues of a 4 week-old mouse were included in the experiment. Lanes 1, 16, and 17 contain yeast tRNA and lane 19 is probe only.
Expression of GKLF was also examined in intestinal tissues obtained from C57BL/6J mice between days 4 (P4) and 210 (P210) of postnatal development. RNA samples from whole small intestine and whole colon were analyzed (Fig. 3). At birth, the level of GKLF transcript is higher in the colon than in the small intestine. The levels in both organs rise gradually with increasing age and peak at 75 days after birth. The overall GKLF transcript level in the colon at each given time point is higher than that in the small intestine because of underloading of the colonic samples as judged by the β-actin levels (compare lanes 6–10 to lanes 1–5, lower panel).
Fig. 3.
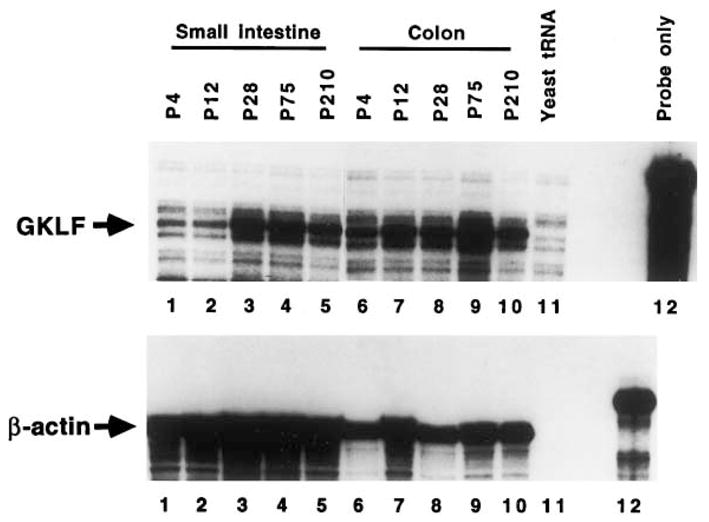
RPA of GKLF transcripts in the intestines during postnatal development. RNA was extracted from whole small intestine or whole colon from mice at the stated days following birth (P4 to P210). Twenty μg of RNA were used in each lane.
Because expression of GKLF has been shown to be associated with growth arrest, we examined its expression in the Min mouse, a model of intestinal tumorigenesis and deregulated proliferation. RPA was performed on RNA obtained from multiple pools of whole small intestine and whole colon of Min mice and compared to wild-type mice between weeks 6 and 15 after birth (Figs. 4 and 5). In the C57BL/6J Min/+ mouse, intestinal tumors are typically first noted at the 9th week after birth and increase in size from then on. These mice usually do not survive beyond 17 weeks of life due to tumor burden [12,17]. The average number of tumors developed in the intestinal tract of closely related pedigrees to our Min/+ mice is consistent with previously published report – 24 in the small intestine and 5 in the colon [17]. As seen in Fig. 4, the level of GKLF transcript is fairly equal in the small intestine of wild-type and Min mice at weeks 6 and 9. However, there is a significant decrease in the GKLF message level in the small intestine of Min mice as compared to the wild-type mice at weeks 12 and 15 (compare lanes 6 to 5 and lanes 8 to 7, Fig. 4). This difference is much less pronounced when RNA from whole colon of wild-type and Min mice were analyzed (Fig. 5).
Fig. 4.
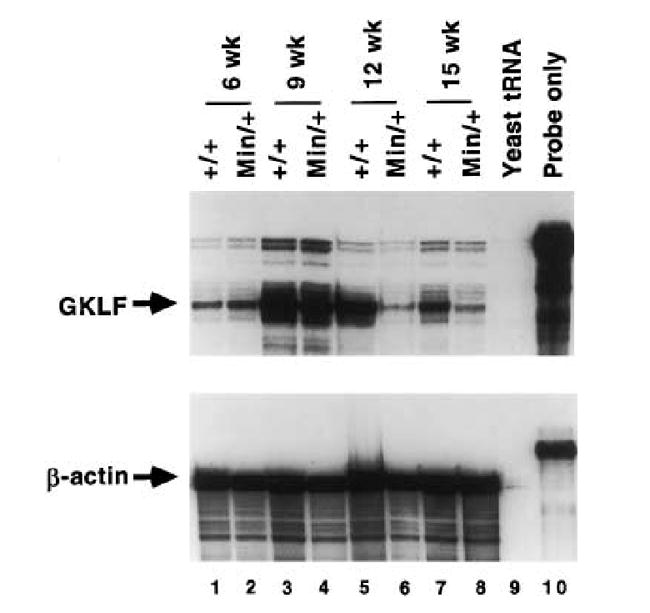
RPA of GKLF transcripts in the small intestine of wild-type and Min mice. RNA was extracted from whole small intestine from wild-type (+/+) or Min (Min/+) mice at the stated weeks after birth. Twenty μg were used in each lane.
Fig. 5.
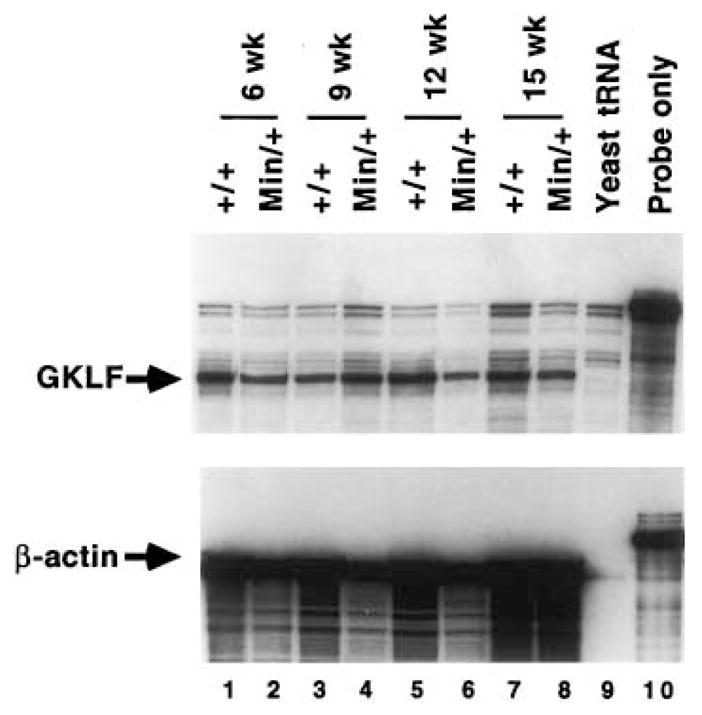
RPA of GKLF transcripts in the colon of wild-type and Min mice. Total RNA was extracted from whole colon of wild-type (+/+) or Min (Min/+) mice at the indicated time points after birth. In this experiment, 10 μg of RNA were used in each lane.
4. Discussion
GKLF was originally identified based on DNA homology to the zinc finger region of a related Krüppel protein, zif268 [14]. The amino acid sequence outside the zinc finger region of GKLF, however, is unique. Of all the Krüppel related proteins, the zinc finger sequences of two transcription factors, erythroid Krüppel-like factor (EKLF [18]) and lung Krüppel-like factor (LKLF [19]), are most similar to that of GKLF. A recent study showed that the nuclear localization signals of these three proteins are also conserved [20]. Each of these proteins is expressed in a tissue-selective fashion, and EKLF has been shown to be an essential factor for hematopoiesis [21,22].
Our previous study showed that GKLF is expressed in a tissue-selective manner, notably at high levels in the gastrointestinal tract [14]. In addition, GKLF is expressed in the skin [15]. Furthermore, in situ hybridization experiments showed that GKLF is specifically expressed in the differentiated epithelial cells of both intestine and skin [14,15]. Combined with the growth arrest-associated nature of GKLF [14], it is possible that GKLF has an important regulatory function during terminal differentiation of specific epithelial cells. These observations provided the rationale for the current study to examine expression of GKLF during development.
Consistent with the hypothesis that GKLF plays a role in regulating epithelial differentiation, we found that GKLF is expressed during a critical period of fetal development when the gut epithelium undergoes major transition from a pseudostratified to a columnar structure between E15 and E18 (Fig. 2). These findings demonstrate a correlation between GKLF expression and an important ‘landmark’ stage of gut epithelial development, which lends support to the hypothesis that GKLF may be involved in regulating intestinal epithelial differentiation.
The decreased expression of GKLF in the small intestine of Min mice during the period of intestinal tumorigenesis is intriguing. However, this is not entirely surprising since GKLF is expressed in growth-arrested cells [14]. As the adenomatous epithelium in the Min intestine is a model of deregulated proliferation, it is possible that the level of GKLF transcript is decreased in the adenomas themselves. The less apparent decrease of GKLF transcript level in the colon of Min mice when compared to the wild-type controls supports this notion since the Min mice develop significantly fewer tumors in the colon as compared to the small intestine [12,17]. It remains to be seen, however, whether the decreased expression of GKLF occurs during the initiation of tumorigenesis or is simply a result of tumor formation due to increased proliferation and/or decreased differentiation.
It is of interest to note that there are several similarities in the pattern of expression of GKLF and the gene encoding another intestine-specific transcription factor, Cdx-2. Cdx-2 is a homeobox-containing nuclear protein specifically expressed in the gut epithelium [23]. In the adult mouse, the level of Cdx-2 transcript is higher in the colon than the small intestine. During embryogenesis, expression of Cdx-2 is significantly up-regulated in the small and large intestine between E16 and E18 [24]. In addition, in both human and rat colorectal adenoma and carcinoma, the level of Cdx-2 protein is markedly reduced [25]. These observations are reminiscent of those for GKLF reported in this and previous studies [14,15]. Furthermore, the importance of Cdx-2 in regulating intestinal epithelial proliferation is demonstrated by a recent study in which mice with targeted ablation of Cdx-2 was shown to develop intestinal polyposis [26]. It remains formally possible that GKLF has a similar function and similar gene targeting experiments may help to clarify the role of GKLF in the control of intestinal epithelial proliferation.
Acknowledgments
We thank Dr. Stanley R. Hamilton for providing the Min mice and age-matched control littermates described in the manuscript. This work was supported in part by grants from the National Institutes of Health (to V.W.Y.). H.T.-T. and J.M.S. were each supported by a National Research Service Award from the National Institutes of Health. C.S.M. was supported by a Student Research Fellowship Award from the American Gastroenterological Association.
Abbreviations
- APC
adenomatous polyposis coli gene
- EKLF
erythroid Krüppel-like factor
- FAP
familial adenomatous polyposis
- GKLF
gut-enriched Krüppel-like factor
- LKLF
lung Krüppel-like factor
- Min
multiple intestinal neoplasia
- nt
nucleotide
- RPA
ribonuclease protection assay
- SI
small intestine
References
- 1.Gordon JI, Hermiston ML. Curr Opin Cell Biol. 1994;6:795–803. doi: 10.1016/0955-0674(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 2.Simon TC, Gordon JI. Curr Opin Genet Dev. 1995;5:577–586. doi: 10.1016/0959-437x(95)80026-3. [DOI] [PubMed] [Google Scholar]
- 3.Wright NA, Irwin M. Cell Tissue Kinet. 1982;15:595–609. doi: 10.1111/j.1365-2184.1982.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman MH. In: The Atlas of Mouse Development. Kaufman MH, editor. Academic Press; San Diego, CA: 1992. [Google Scholar]
- 5.Traber PG, Silberg DG. Annu Rev Physiol. 1996;58:275–297. doi: 10.1146/annurev.ph.58.030196.001423. [DOI] [PubMed] [Google Scholar]
- 6.Calvert R, Pothier P. Anat Rec. 1990;227:199–206. doi: 10.1002/ar.1092270208. [DOI] [PubMed] [Google Scholar]
- 7.Henning SJ. Annu Rev Physiol. 1985;47:231–245. doi: 10.1146/annurev.ph.47.030185.001311. [DOI] [PubMed] [Google Scholar]
- 8.Burt RW, Lipkin M. In: The Genetic Basis of Common Diseases. King RA, Rotter JI, Motulsky AG, editors. Oxford University Press; New York, NY: 1992. pp. 650–680. [Google Scholar]
- 9.Bussey HJR. In: Familial Polyposis Coli: Family Studies, Histopathology, Differential Diagnosis, and Results of Treatment. Bussey HJR, editor. Johns Hopkins University Press; Baltimore, MD: 1975. [Google Scholar]
- 10.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, Finniear R, Markham A, Froffen J, Boguski MS, Altschul SF, Horaii A, Ando H, Kyoshi Y, Miki Y, Nishisho I, Nakamura Y. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 11.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P, Markham A, Krush AJ, Peterson G, Hamilton SR, Nilber DB, Levy MC, Bryan TM, Preisinger AC, Smith KJ, Su LK, Kinzler KW, Vogelstein B. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 12.Moser AR, Pitot HC, Dove WF. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 13.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 14.Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 16.Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 17.Moser AR, Dove WF, Roth KA, Gordon JI. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller IJ, Bieker JJ. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson KP, Kern CB, Crable SC, Lingrel JB. Mol Cell Biol. 1995;15:5957–5965. doi: 10.1128/mcb.15.11.5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shields JM, Yang VW. J Biol Chem. 1997;272:18504–18507. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 22.Perkins AC, Sharpe AH, Orkin SH. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 23.James R, Kazenwadel J. J Biol Chem. 1991;266:3246–3251. [PubMed] [Google Scholar]
- 24.James R, Erler T, Kazenwadel J. J Biol Chem. 1994;269:15229–15237. [PubMed] [Google Scholar]
- 25.Ee HC, Erler T, Bhathal PS, Young GP, James RJ. Am J Pathol. 1995;147:586–592. [PMC free article] [PubMed] [Google Scholar]
- 26.Chawengsaksophak K, James R, Hammond VE, Köntgen F, Beck F. Nature. 1997;386:84–87. doi: 10.1038/386084a0. [DOI] [PubMed] [Google Scholar]