

SUMMARY
Constitutive egress of bone marrow (BM)-resident hematopoietic stem and progenitor cells (HSPCs) into the blood is a well-established phenomenon, but the ultimate fate and functional relevance of circulating HSPCs is largely unknown. We show that mouse thoracic duct (TD) lymph contains HSPCs that possess short- and long-term multilineage reconstitution capacity. TD-derived HSPCs originate in the BM, enter the blood and traffic to multiple non-lymphoid extramedulary tissues, where they reside for at least 36h until entering draining lymphatics to return to the blood and, eventually, the BM. HSPC egress from extramedullary tissues into lymph depends on sphingosine-1-phosphate (S1P) receptors, particularly S1P1. Migratory HSPCs proliferate within extramedullary tissues giving rise to tissue-resident myeloid cells, preferentially dendritic cells. HSPC differentiation is amplified upon exposure to Toll-like receptor agonists. Thus, HSPCs can survey peripheral organs and replenish tissue-resident hematopoietic cells by acting as a source of specialized leukocytes during host defense against pathogens.
INTRODUCTION
Most differentiated cells found in mammalian blood have variable but limited life spans and must be constantly replenished. Blood cell homeostasis depends on a rare population of precursor cells, the hematopoietic stem cells (HSCs), which possess the unique capacity for self-renewal and multilineage differentiation. The function of HSCs and the partially lineage-committed progenitor cells that arise from them has been linked to their migratory properties, at least during fetal life when the anatomic seat of hematopoietic activity changes several times (Cumano and Godin, 2007).
In postnatal mammalian life HSPCs reside mostly in specialized niches in bone marrow (BM) cavities that control HSPC survival, proliferation, self-renewal, and differentiation (Adams and Scadden, 2006). However, even in adulthood HSPCs are not entirely sessile, but contain a population of highly migratory cells. It is well established that some HSPCs recirculate constantly between BM and blood (Goodman and Hodgson, 1962; Wright et al., 2001b). Accordingly, normal blood from adult mice contains a small, but stable population of several hundred HSPCs, which upon transplantation to irradiated recipients are capable of long-term reconstitution (LTR) of hematopoietic activity (Fleming et al., 1993; Morrison et al., 1997). It has been speculated that the continuous trafficking of HSPCs between BM and blood is a mechanism to maintain full occupancy of HSPC niches in all BM cavities (Wright et al., 2001b). However, the exact trafficking pathways of blood-borne HSPCs and the physiological relevance of their postnatal migration remain largely unclear.
The daily turnover of HSPCs that enter and leave the bloodstream is believed to be high (Wright et al., 2001b). The BM is probably not the exclusive physiological source and destination of blood-borne HSPCs, because HSPCs have also been recovered from extramedullary sites, like the liver (Cardier and Barbera-Guillem, 1997), spleen (Wright et al., 2001b), and muscle (McKinney-Freeman et al., 2002). Therefore, although we know little about the migratory dynamics of extramedullary HSPCs, it seems likely that circulating HSPCs visit anatomic regions other than the BM. A case in point is the trafficking of mature lymphocytes, which extravasate continuously into multiple lymphoid and non-lymphoid tissues. Most tissue-resident lymphocytes eventually return to the blood via lymphatics that drain into the thoracic duct (TD). This lymphocyte recirculation is essential for immunosurveillance because it maximizes the probability that lymphocytes encounter rare cognate antigens (von Andrian and Mackay, 2000).
Here, we have examined whether blood-borne HSPCs might follow similar extramedullary traffic patterns as lymphocytes. We demonstrate that efferent lymphatics contains a stable fraction of HSPCs that possess short- and long-term multilineage reconstitution capacity. TD HSPCs originate in the BM and traffic constitutively to multiple extramedullary, non-lymphoid tissues where they reside for at least 36h until entering the draining lymphatics to return to the blood. This recirculation of HSPCs is regulated, in part, by the S1P receptor S1P1 and may foster the local production of tissue-resident innate immune cells under both steady-state conditions and in response to infections.
RESULTS
Lin− hematopoietic cells travel in the TD
We surmised that if HSPCs recirculate through extramyeloid tissues then they, like differentiated lymphocytes, might become lymph-borne. Indeed, lymph fluid collected from murine TD (see supplemental methods) contained up to 4% mononuclear cells (MNCs) that expressed the pan-leukocyte antigen CD45 but no other hematopoietic lineage markers (Fig. 1A). This population included Lin−IL-7Rα+c-Kit+Sca-1+ (∼0.003-0.004% of all TD-MNCs) and Lin−IL-7Rα−c-Kit+Sca-1− cells (∼0.01-0.03% of all TD-MNCs), resembling the phenotype of committed BM common lymphoid (CLP; (Kondo et al., 1997)) and common myeloid progenitor cells (CMP; (Akashi et al., 2000)), respectively (suppl. Fig. 1). In addition, CD45+ Lin−IL-7Rα-TD cells included cells with a primitive HSC/multi-potent progenitor (MPP) phenotype (Lin−IL-7Rα−c-Kit+Sca-1+ (LSK)) (Christensen and Weissman, 2001), which amounted to 0.001-0.004 % of CD45+ TD-MNCs (Fig. 1A and suppl. Fig. 1). Lymph-derived LSK cells expressed several adhesion/traffic molecules, including CXCR4, CD44, PSGL-1, LFA-1, as well as α4 and α5 integrins (supplemental table). Methylcellulose-based colony-forming unit culture (CFU-C) assays of MNCs from blood and TD confirmed that both compartments contain clonogenic HSPCs (Fig. 1B). Notably, no significant differences were observed in the number of CFU-Cs in peripheral blood collected 6, 12, or 24 hrs after surgery or with control blood (without surgery), implying that the collection of thoracic duct lymph per se did not induce enhanced mobilization of HSPCs (data not shown). Clonogenic cells mostly resided within the Lin−c-Kit+ population of TD cells, whereas Lin+ or Lin−c-Kit− TD cells gave rise to no or few colonies in CFU-C assays, respectively (Fig. 1C). When lymph was collected from irradiated wildtype (WT) mice reconstituted with BM from GFP-transgenic donors (Wright et al., 2001a) virtually all colonies expressed GFP, indicating that the lymph-borne HSPCs are BM-derived (suppl. Fig. 2A). On average, we measured a TD lymph flow of 0.78 ± 0.05 ml/h and each ml of lymph contained 4.9 ± 0.8 CFU-C. Considering that TD lymph constitutes only ∼50% of total body lymph flow we estimate that ∼200 clonogenic HSPCs pass through the lymph of a mouse every day. This number is substantial considering the fact that a single HSC can reconstitute the entire hematopoietic system of a mouse (Wagers et al., 2002) and that infusion of as few as 30 of these cells is sufficient to save 50 percent of lethally irradiated mice, and to reconstitute all blood cell types in the survivors (Spangrude et al., 1988).
Figure 1. Clonogenic HSPCs travel in TD lymph.

A, FACS analysis of TD MNCs. Numbers show the frequency of gated cells among total MNCs. No events where detected in the LSK gate with isotype-matched control antibodies (data not shown). B, Concentration of CFU-Cs in normal mouse PB and TD lymph. Columns and error bars represent mean±sem, n=12-23 assays (from 6-12 animals), circles represent results from individual assays. C, Absolute (left) and relative frequencies (right) of CFU-Cs in total MNCs or sorted Lin+, Lin−c-Kit−, and Lin−c-Kit+ TD cells. Mean±sem are presented, n=4-16 assays. Right panel: Representative micrographs of Lin-c-Kit+ TD-derived CFU-Cs, defined by the presence of granulocytes, macrophages, erythroid cells and megakaryocytes as CFU-granulocyte (CFU-G), CFU-macrophage (CFU-M), CFU-granulocyte, macrophage (CFU-GM), CFU-granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM), respectively. Bars represent 50μm.
Lin− TD cells have BM homing capacity
To define whether lymph-borne Lin− cells share the ability of blood-derived HSPCs to home to the BM, fluorescently tagged Lin− TD cells were injected i.v. into mice and their presence in recipient tissues was assessed by flow cytometry 24h later. Irrespective of whether recipients were preconditioned by irradiation, donor Lin− cells were detectable in recipient BM and spleens (suppl. Fig. 2B). Multiphoton intravital microscopy of skull BM (Mazo et al., 2005) in anesthetized recipients confirmed that Lin− TD-derived cells can migrate into BM cavities within 12-24h after adoptive transfer (suppl. Fig. 2C, supplemental movies 1-3).
HSPCs with short- and long-term multilineage reconstitution capacity travel in the TD lymph
To explore the functional engraftment capacity of lymph-borne HSPCs, TD-MNC from CD45.2+ donors were transplanted into sublethally irradiated CD45.1+ congenic recipients. After 6 weeks, ∼15% of Gr-1+ peripheral blood granulocytes were donor-derived (Fig. 2A), indicating that lymph-borne HSPCs possess myeloid differentiation potential (Morrison and Weissman, 1994; Wright et al., 2001b). To test whether they also give rise to T cells, we harvested TD-MNCs from athymic B6nude/nude mice, which lack mature T cells, although their HSPCs can give rise to T cells when transplanted to thymus-sufficient animals. B6nude/nude TD-MNCs were injected into lethally irradiated Rag2−/−γc−/− mice, which possess a functional thymic anlage but are genetically incapable of generating lymphocytes (Goldman et al., 1998). Remarkably, six weeks after transplantation Rag2−/−γc−/− recipients of B6nude/nude TD-MNCs had a near-normal T cell compartment (Fig. 2B). To address whether lymph-borne HSPCs contain multipotent HSCs with persistent self-renewal capacity, CD45.2+ TD-MNCs were transplanted into lethally irradiated CD45.1+ recipients, which were sacrificed 12 weeks later. CD45.2+ cells were then sorted from the first recipients' BM and transplanted into another CD45.1+ recipient (Fig. 2C). Ten weeks after the secondary transplantation, the recipient showed multilineage reconstitution by CD45.2+ cells. Hence, normal murine lymph contains long-term multilineage repopulating HSPCs that meet the phenotypic and functional criteria of true HSCs.
Figure 2. TD lymph HSPCs possess long-term multilineage repopulation capacity.
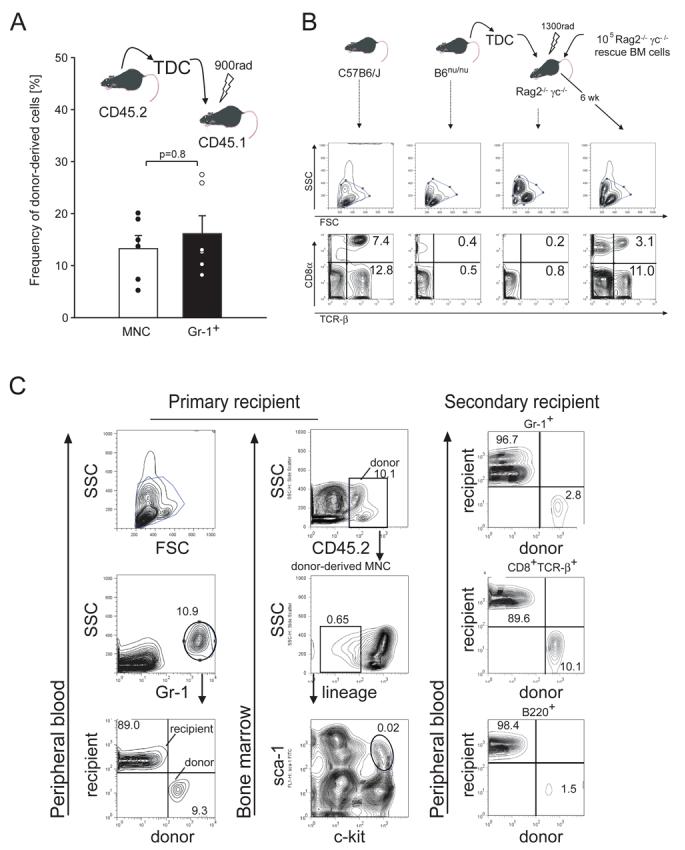
A, TD-derived CD45.2+ MNCs (2 × 107) were injected into sublethally irradiated CD45.1+ recipients. Chimerism among total MNC and Gr-1+ cells in PB was determined by FACS 6wks after transplantation, n=6. B, TCR-β+ T cells are found in PB of WT mice (first column), but not in nude (second column) or Rag2−/−γc−/− mice (third column). Transfer of TD-MNCs (2×107) from nude mice into lethally irradiated Rag2−/−γc−/− mice restored T cells in Rag2−/−γc−/− recipients (fourth column). Numbers show the frequency of CD8α+ TCR-β+ and CD8α− (mostly CD4+) TCR-β+ cells among MNCs. Data are representative of 2 experiments. C, CD45.2+ TD-MNCs (2×107) were transplanted into sublethally irradiated CD45.1+ primary recipients. Twelve weeks later, chimerism among PB granulocytes (Gr-1+) was determined (left column). Analysis of CD45.2+ cells in BM (middle column) demonstrates the persistence of a Lin− subset, which includes LSK cells (indicated by circle in bottom panel). Right column: Twelve weeks after primary transplantation, the CD45.2+ CD45.1− cells were sorted out of the primary recipient's BM and transplanted into a lethally irradiated CD45.1+ secondary host. Chimerism among PB granulocytes (Gr-1+), T cells (CD8+TCR-β+), and B cells (B220+) was assessed by FACS 12 weeks after secondary transplantation. Numbers show the frequency of cells in the individual quadrants.
HSPCs do not require lymphoid structures for recirculation
HSPCs travel between blood and lymph similar to naive lymphocytes. Hence both populations might share transit routes guiding them from blood into lymphatics. Naive lymphocytes must home to lymph nodes and Peyer's patches (PPs) to gain access to lymph vessels (von Andrian and Mackay, 2000). Consequently, MNCs were dramatically reduced in lymph of lymphotoxin (LT)-α deficient mice (Fig. 3A), which lack these lymphoid tissues (De Togni et al., 1994). However, HSPCs were still present in the TD lymph of LT-α−/− mice as assessed by FACS (data not shown) or CFU-C assays (Fig. 3B). Thus, HSPCs, unlike lymphocytes, do not require secondary lymphoid organs (SLOs) to recirculate, but might traffic directly to the lymph from extramedullary non-lymphoid tissues.
Figure 3. Secondary lymphoid organs are not required for recirculation of HSPCs across extramedullary, non-lymphoid tissues.

CFU-C assays were performed using TD-MNC collected from WT or LT-α−/− mice. A, concentration of MNC in lymph, n=6-12 animals; B, relative (left) and absolute frequencies (right) of CFU-Cs among lymph-borne MNCs, n=14-28 assays. Bars show mean±SEM.
HSPCs continuously travel from the BM to various extramedullary tissues
To define where and how HSPCs might leave the blood and pass through extramedullary tissues into the lymph, we first surveyed different murine tissues for the presence of HSPCs using CFUC assays. Clonogenic progenitors were detectable in many tissues, including the lung, liver, small intestine, kidneys and blood, but CFU-Cs were rare or absent in cardiac and skeletal muscle, skin and LNs (suppl. Fig. 3). It should be noted that we counted the number of colonies 7 days after plating of the cells in methylcellulose to detect primitive HSPCs with large burst size. Therefore, our data do not account for the presence of late-outgrowth (more committed) progenitors, that have been reported to reside within some tissues including peripheral LNs (MacVittie and McCarthy, 1977). Virtually all clonogenic tissue HSPCs were BM-derived, since colonies uniformly expressed GFP in chimeric wildtype recipients of GFP+ BM (data not shown). Moreover, transplantation of liver-derived CFU-Cs into irradiated recipients resulted in multilineage reconstitution, indicating that at least in some organs, clonogenic cells possess multilineage reconstitution capacity (data not shown). Based on the yield of CFU-Cs in surveyed organs, we estimate the total number of HSPCs resident in extramedullary non-lymphoid tissue to be at least twice as large as the average number of HSPCs circulating in murine blood (Wright et al., 2001b).
Next, we generated pairs of parabiotic mice, in which one partner ubiquitously expressed GFP. Consistent with previous reports (Wright et al., 2001b), cross-circulation was established by day 3 after surgical joining, and the host:partner (i.e GFP+:GFP−) ratio of both mature leukocytes and CFU-C in blood plateaued at ∼3:2 (corresponding to 40% chimerism) between days 10 and 14 (not shown). In contrast, CFU-C chimerism in the BM reached only ∼5% after 14 days of parabiosis (Fig. 4A). Similar levels of BM chimerism were reported in mice joined for up to 16 weeks (Wagers et al., 2002). In extramedullary tissues of parabiotic mice, the highest level of HPC chimerism was in the spleen (40±7%), followed by the lungs (31±6%), liver (24±9%) and kidneys (15±5%), implying that within these organs tissue-resident HSPCs are constantly replenished by blood-borne HSPCs and are turned over within a matter of days. Partner-derived CFU-Cs were undetectable in the small intestine or brain. We also detected marked CFU-C chimerism (27±1%) in lymph of parabiotic mice, indicating that at least some homed HSPCs are not permanently retained in extramedullary tissues, but recirculate freely between lymph and blood (Fig. 4B).
Figure 4. Recirculation of HSPCs through multiple extramedullary, non-lymphoid tissues.

A, WT and β-actin/GFP transgenic mice were parabiosed and analyzed by isolating MNCs from indicated tissues and testing for GFP+ and GFP− CFU-C content (bar graph). Micrographs show representative brightfield (left) and fluorescence (right) images of host- and partner-derived CFU-Cs. *P<0.05, n=2-8. B, CFU-C chimerism in TD lymph of parabiotic mice, n=4. C, CFU-C chimerism in PB (closed circles) and TD lymph (open circles) at different time points after surgical separation of parabiotic mice, n=4-8 experiments.
To define the kinetics at which HSPCs enter the lymph after having accessed extramedullary tissues, some pairs of mice were surgically separated after 14 days of parabiosis. Following separation, blood chimerism decreased rapidly, while lymph-borne HSPC chimerism was unchanged during the first 36h and declined only thereafter (Fig. 4C). Given this delayed drop in HSPC chimerism, we estimate that the mean dwell-time of HSPCs that have homed to peripheral tissues is at least 36h. This suggests that many organs are sites of active HSPC traffic from blood to tissue to lymph, but certain organs recruit few, if any, circulating HSPCs, at least over the two-week time interval tested. Of note, a tracer (1% Evans Blue) injected directly into femur BM did not appear in any juxtaposed LN (not shown), but became rapidly visible in draining LNs after intracutaneous injection. This is consistent with the long-held notion that mammalian BM lacks lymphatic drainage. Thus, HSPCs in the BM egress directly into the blood, whereas lymph-borne HSPCs have most likely departed from tissues other than the BM.
Sphingosine 1-phosphate (S1P) and S1P receptor S1P1 regulate the egress of HSPCs from peripheral tissues into draining lymphatics
Next, we sought to determine whether chemoattractants guide HPC recirculation. Most chemoattractants that control leukocyte migration signal through Gαi-coupled receptors that can be inhibited by pertussis toxin (PTX). Indeed, as early as 2-12h after a single PTX injection we observed a dramatic drop in the number of lymph-borne CFU-Cs (Fig. 5A). In blood, the number of CFU-Cs was not significantly altered during this time interval, however, their number increased markedly at 24-96h after PTX injection ((Papayannopoulou et al., 2003) and data not shown). Thus, Gαi-coupled chemoattractants might play a role in the long-term recruitment of HSPCs from blood to tissues. However, our parabiosis experiments predict that the dwell time of HSPCs within tissues is >36h. This implies that a PTX-induced drop in lymph-borne CFU-Cs should only become manifest after ∼36h if migration across the blood-tissue interface is the only step affected by PTX. The much more rapid time-course of PTX-induced CFU-C disappearance from lymph implies an essential role for a Gαi-coupled signal that directs tissue-resident HSPCs into draining lymphatics.
Figure 5. Egress of HSPCs from extramedullary tissues into lymph is PTX-sensitive and depends on S1P. A, Effect of PTX on lymph-borne CFUc.

Lymph was collected for 8h from mice that were previously treated for 2h with vehicle or 1μg PTX, and CFU-C concentration was measured. Bars show mean±SEM, n=4-6 assays. B, Effect of FTY720 on lymph-borne MNCs and CFUc. Effect of treatment with S1P receptor antagonist FTY720 (6 hr) on MNC concentration (left panel, n=4-7) and CFU-C content (right panel, n=12-14) in lymph. C, Effect of FTY720 in LTx-α−/− mice. CFU-C assays were performed using thoracic duct MNC collected from LT-α deficient mice, treated with vehicle or FTY720. Lymph MNCs were collected for 8h from FTY720-pretreated mice starting at 6 or 48h after the last drug administration. The number of CFU-Cs per ml of lymph is shown. Error bars show s.e.m. D, Effect of FTY720 on blood-borne MNCs and CFUc. Effect of treatment with FTY720 (6hr) on MNC concentration (left panel) and CFU-C frequency (right panel) in PB. E, Effect of FTY720 on hepatic CFUc. Liver MNCs were collected from mice treated with vehicle or FTY720 (3mg/kg) for 7 days. Total number of CFU-C per liver are given. F, Effect of THI on lymph-borne CFUc. CFU-C content in TD lymph of control mice (vehicle) and animals that had received the S1P lyase inhibitor THI for three days (n=7-8).
Egress of tissue-resident lymphocytes into lymphatics is controlled by S1P1, a Gαi-coupled receptor for S1P (Massberg and von Andrian, 2006; Rosen and Goetzl, 2005). We found that murine HSPCs express S1P1, as well as S1P2, S1P3, and S1P4 mRNA (suppl. Fig. 4A). Although quantitative real-time PCR showed that the S1P1 mRNA expression levels are lower on HSPC subsets compared to B and T cells (suppl. Fig. 4B), they were much higher than on myeloid cells, and both progenitor subsets migrated toward S1P in vitro (suppl. Fig. 4C). Thus, we asked whether S1P signalling might influence HSPC recirculation. Most S1P receptors, including S1P1, are blocked by the immunosuppressant FTY720, which induces lymphocyte sequestration in SLOs causing profound lymphopenia in blood and lymph (Chiba et al., 1998; Mandala et al., 2002; Matloubian et al., 2004). Remarkably, treatment of mice with FTY720 for 6h nearly abolished lymph-borne CFU-Cs (Fig. 5B). Among the multiple S1P receptors expressed by HSPCs, S1P1 appeared to play the predominant role for this effect, since SEW2871, which selectively targets S1P1 (Jo et al., 2005), transiently reduced the appearance of lymph-borne CFU-Cs to a similar extent as FTY720 (suppl. Fig. 4D). Disappearance of lymph-borne CFU-Cs in response to S1P receptor inhibition did not require HSPC sequestration in SLOs, since FTY720 blocked HSPC recirculation also in LT-α−/− mice (Fig. 5C). HSPCs reappeared in the lymph of LT-α−/− mice within two days after discontinuation of FTY720-treatment (Fig. 5C), suggesting that FTY720 does not affect the viability of HSPCs. Likewise, FTY720 did not alter the capacity of HSPCs to form colonies in CFU-C assays (data not shown).
Notably, FTY720 not only depleted lymph-borne HSPCs but also induced a significant (74±5%) drop of CFU-Cs in the blood (Fig. 5D). Based on our parabiosis experiments, a decrease of HSPC migration across the blood-tissue interface would translate into a decrease in lymph CFU-Cs only after ∼2 days. This suggests that the drop of blood CFU-Cs alone cannot account for the disappearance of CFU-Cs from the lymph observed within 6h after FTY720-treatment. Treatment of mice with FTY720 over 7 days resulted in a significant increase in the number of HSPCs residing within extramedullary tissues (Fig. 5E). Hence, our data imply that S1P receptors, particularly S1P1, regulate the egress of HSPCs from tissue into the draining lymphatic vasculature. The number of HSPCs in the BM of FTY720-treated mice was not significantly altered as compared to vehicle-treated animals (suppl. Fig. 4E). Hence FTY720-induced disappearance of HSPCs from blood was due to inhibition of HSPC recirculation from extramedullary tissues. Consistent with this notion, we did not observe a significant difference in the number of HSPCs mobilized to the blood in response to G-CSF between mice treated with FTY720 or vehicle (data not shown).
S1P is abundant in blood and lymph, but low in lymphoid and non-lymphoid tissues due to rapid interstitial degradation by tissue-resident S1P lyase (Schwab et al., 2005). Extravascular lymphocytes in LNs enter the lymph in response to steep S1P gradients. This egress step can be inhibited by pharmacological blockade of S1P lyase with 2-acetyl-4-tetrahydroxybutylimidazole (THI) (Schwab et al., 2005). To test whether S1P degradation through S1P lyase is also required for HSPC recirculation, we collected lymph from THI-treated mice. As reported previously (Schwab et al., 2005), THI treatment resulted in profound lymphopenia in blood and lymph (data not shown). Concomitantly, lymph-borne HSPCs were markedly reduced (Fig. 5F), indicating that S1P lyase is required for HSPC passage through tissues.
Tissue-resident HSPCs give rise to myeloid cells in peripheral tissues
To test whether recirculating HSPCs might give rise to mature hematopoietic cells locally within extramedullary tissues, we implanted HSPC-enriched Lin−GFP+ TD-derived cells (5×105) or HSC-enriched BM-derived Lin−c-Kit+Sca-1+ cells (104) under the left kidney capsule of recipient mice (Fig. 6A and B). Medium without cells was injected into the contralateral (right) kidney. Six days later, a sizeable number of GFP+ cells was detected within the kidney implanted with Lin−GFP+ TD cells and, especially, after implantation of HSC-enriched BM-derived Lin−cKit+Sca-1+ cells (Fig. 6A, B). A significantly lower but stable number of GFP+ cells was observed in the medium-injected control kidney (P<0.05, Fig. 6A), but also within the blood and the BM as assessed by CFU-C assay after implantation of Lin−c-Kit+Sca-1+ cells (Fig. 6C). Confocal microscopy showed that virtually all GFP+ cells in injected kidneys co-expressed the pan-hematopoietic marker CD45 (suppl. Fig. 5A). The majority (> 50%) of the GFP+ cells co-expressed myeloid lineage markers (Fig. 6A and suppl. Fig. 5A, B) and ∼10% co-expressed B220, while no CD3 (T cell lineage) expressing GFP+ cells were detected (suppl. Fig. 5A). The absolute numbers of GFP+ cell with myeloid phenotype were 26- and 14-fold higher in the kidneys injected with Lin−GFP+ or Lin−c-Kit+Sca-1+GFP+ cells, respectively, compared to the contralateral cell-free medium-treated kidneys. This implies that, after deposition within tissues, HSPCs have the capacity to differentiate locally into myeloid lineages. Cell fusion is not likely to account for this process, as co-expression of CD45.1 and GFP was never observed when GFP+ CD45.2+ HSPC-enriched cells were implanted into CD45.1+ kidney capsules (data not shown).
Figure 6. HSPCs proliferate and give rise to myeloid cells in peripheral tissues particularly in response to TLR ligands.

A-B, Distinct TD- or BM-derived HSPC-enriched populations were implanted under the left kidney capsule of WT mice with medium or LPS with or without preincubation for 12h as indicated. Medium or LPS without cells was injected under the contralateral control kidney capsule. A, Quantitative analysis of the total number of GFP+ cells (G) and of GFP+ myeloid cells (M) in the left (empty bars and filled circles) and in the right (control, open circles) kidney. *P<0.05 vs. control kidney, #P<0.05 vs. medium, n=3 experiments, mean±SEM. B, Representative confocal images showing expression of GFP and reactivity with a mixture of mAbs to myeloid markers (CD11c, CD11b, F4/80, Gr-1) 6 days after implantation. Bars represent 50μm. C, Detection of GFP+ (donor-derived) CFU-C within the BM (left) and PB (right) after implantation of Lin-c-Kit+Sca-1+ cells preincubated with medium or LPS for 12h. Mean±SEM, n=3 experiments. D-E, 104 HSPC-enriched Lin−c-Kit+ GFP+ cells were implanted into the left kidney capsule of WT recipient mice together with LPS. D, Representative micrographs showing the expression of GFP, Gr-1, CD11c, or the PDC marker 120G8 in the kidney 6 days later. Bars represent 50μm. E, Relative subset frequencies among GFP+ cells in serial kidney sections, n=3 experiments, mean±SEM. F, Representative confocal images showing expression of GFP, incorporation of BrDU and expression of Ki-67 6 days after implantation of the indicated HSPC populations. Bars represent 10μm. Arrows indicate GFP+ BrDU+ cells, arrow heads show GFP− BrDU+ cells.
Tissue-resident HSPCs respond to TLR ligands
HSPCs express TLRs and their co-receptors, including TLR4, MD-2 and CD14, required for recognition of bacterial lipopolysaccharide (LPS) from Gram-negative bacteria (Nagai et al., 2006). In vitro, TLR signaling drives differentiation of HSPCs into myeloid lineages. Migratory HSPCs which encounter TLR ligands within extramedullary tissues might thus act as a highly versatile local source to rapidly replenish innate immune cells during infection. We therefore incubated distinct subsets of TD- or BM-derived HSPC-enriched cell populations for 12h with LPS (10μg/ml) and implanted them with LPS underneath the left kidney capsule. LPS (10μg/ml) without HSPCs cells was injected into the contralateral (control) kidney (Fig. 6A, B). Six days after implantation large numbers of GFP+ cells were present in all kidneys injected with GFP+ HSPCs subsets (Fig. 6A, B). These cells were typically located in clusters, each comprising up to ∼400 cells. Similar findings were obtained when we instilled LPS beneath the kidney capsule immediately prior to implantation of HSPCs that were not previously incubated with LPS (Fig. 6A). The highest numbers of GFP+ cells were observed after implantation of HSC-enriched Linc−Kit+sca-1+ cells (104), whereas HSC-depleted Lin−c-Kit+sca-1− cells (enriched in CMPs) gave rise to comparably lower numbers of GFP+ cells.
In the presence of LPS more than 95% of the GFP+ cells co-expressed myeloid lineage markers (Fig. 6A and B). Most cells (>70% of all GFP+ cells) expressed the dendritic cell marker CD11c (Fig. 6D, E and suppl. Fig. 5C). A small subset (∼10%) of GFP+ cells expressed Gr-1 at intermediate to high levels, indicative of monocyte/macrophage and/or granulocyte differentiation, while very few (<1%) GFP+ myeloid cells were stained with 120G8 mAb, which recognizes plasmacytoid dendritic cells (Fig. 6D, E and suppl. Fig. 5C and data not shown). Most of the GFP+ clusters contained either CD11c+ or Gr-1+ cells, while <10% of the clusters contained a mixture of both subsets. A sizeable fraction (>15%) of GFP+ cells expressed neither CD11c nor Gr-1, suggesting that some cells remained either undifferentiated or gave rise to other leukocyte subsets.
In all instances, the numbers of all GFP+ cells and of myeloid GFP+ cells were significantly higher after implantation of HSPCs together with LPS compared to kidneys injected with the same subset of GFP+ HSPCs without LPS (Fig. 6A), suggesting their local proliferation after TLR ligand engagement. When recipient mice were treated with BrDU after implantation of HSPC-enriched GFP+ cells, the majority of the cells of each of the GFP+ clusters had incorporated BrDU into their nucleus (Fig. 6F). Similar findings were obtained when clusters were stained for the proliferation marker Ki-67 (Fig. 6F), implying that in the presence of TLR ligands HSPCs proliferate locally. Engagement of LPS also reduced the exit of clonogenic HSPCs out of peripheral tissues, since the number of donor-derived GFP+ CFU-Cs was significantly reduced in both peripheral blood and BM following implantation of LPS-pretreated cells (Fig. 6C). Along these lines, the number of GFP+ cells in the control kidneys (injected with medium or LPS without cells), was significantly higher in mice injected with medium as compared to LPS treated mice. Although the overall number of donor-derived cells was low in the control kidneys injected with LPS without HSPCs, we detected GFP+Ki-67+ myeloid clusters in these kidneys (Fig. 6F), providing additional in vivo evidence for the concept of HSPC migration to peripheral tissues contributing to myeloid defense in inflamed organs.
LPS interferes with S1P-S1P1 signalling in recirculating HSPCs
Finally we asked whether TLR stimulation might modulate the capacity of HSPCs to migrate toward S1P gradients. We sorted Lin− or Lin−c-Kit+ TD cells and measured S1P1 mRNA levels following incubation with LPS or medium for 12h. Compared with medium-treated cells, TD-derived Lin− and Lin−c-Kit+ TD cells that had been exposed to LPS, had 2-3-fold reduced S1P1 transcript levels (Fig. 7A, P<0.05). Down-regulation of S1P1 in response to LPS was paralleled by failure of LPS-treated HSPCs to chemotax toward gradient of S1P, while migration toward CXCL12/SDF-1α was enhanced (Fig. 7B). Hence LPS recognition not only triggers local proliferation and differentiation, but also leads to retention of HSPCs within extramedullary tissues (Fig. 6C), at least in part by interfering with S1P-S1P1 dependent signaling.
Figure 7. A TLR ligand reduces the capacity of HSPCs to migrate to sphingosine 1-phosphate.

A, Quantitative PCR analysis of S1P1 mRNA expression by medium- or LPS-treated Lin− or Lin−c-Kit+ TD-derived cells. The amount of S1P1 mRNA is expressed as relative amount of LPS-treated cells normalized to GAPDH. Experiments were performed in quadruplicate, *P<0.05. B, Lin− BM-derived cells pretreated for 12h with LPS or medium were tested for their ability to migrate to 100nM S1P or to 50nM CXCL12/SDF-1α. Migration to the lower chamber was determined by flow cytometry and is given as percentage of the number of input cells; n=3 experiments per group, *P<0.05 vs. SDF-1α. C, Schematic model illustrating the trafficking of migratory HSPCs under physiological conditions and during ainflammation (for details see text).
DISCUSSION
Our data suggest that extramedullary tissues are constitutively surveyed by a pool of recirculating hematopoietic progenitors that are highly versatile and can respond rapidly to distress signals from the local microenvironment (Fig. 7). Naive T and B lymphocytes, another population of recirculating cells, gain access to the lymphatic vasculature after homing to SLOs, which they reach through specialized postcapillary venules, the HEV (von Andrian and Mackay, 2000). However, HSPCs can be recovered from TD lymph of lymphotoxin-α deficient mice that lack HEVs and SLOs (De Togni et al., 1994). This implies that blood-borne HSPCs do not require SLOs to access lymphatics. Accordingly, we recovered few, if any, CFU-Cs from pooled LNs. Although HSPCs entering the lymph in peripheral non-lymphoid tissues must pass through at least one draining LN before reaching the TD, lymph-borne HSPCs that give rise to early CFU-Cs (within 7 days of culture) are apparently not retained in the LN stroma but continue to travel through efferent lymph conduits into the blood. This lack of HSPC accumulation in LNs is most likely due to the fact that HSPCs lack CCR7 (Wright et al., 2002), which is essential for homing to LNs (Debes et al., 2005; Forster et al., 1999; Stein et al., 2000). However, HSPCs express multiple other traffic molecules, including PSGL-1, VLA-4 (CD49d/CD29), LFA-1 (CD11a/CD18), CD44, and the chemokine receptor CXCR4, which are likely to contribute to HSPC migration to non-lymphoid tissues, particularly sites of inflammation (supplemental table and (Wright et al., 2002)).
While the recruitment mechanism(s) responsible for HSPC trafficking from blood into extramyeloid tissues remain(s) to be elucidated, our experiments have uncovered a pivotal role for S1P during the subsequent departure of tissue-resident HSPCs into lymphatics. S1P plays a key role in enabling lymphocyte egress from the thymus into the blood and from SLOs into the lymph (Cyster, 2005; Massberg and von Andrian, 2006; Rosen and Goetzl, 2005). Our study demonstrates that S1P and its receptors, in particular S1P1, also regulate HSPC recirculation. HSPCs disappeared from blood and TD lymph when WT or lymphotoxin-α deficient mice were treated with FTY720. In principle, this effect would be expected with any treatment that blocks HSPC trafficking from one compartment to another, be it from the BM to the blood, from blood to extramyeloid tissues, or from tissues into the lymph. However, our parabiosis experiments revealed that HSPCs need at least 36h to transit from the blood across peripheral tissues into the draining lymph, so treatment effects that block HSPC homing from blood to tissues would only result in reduced lymph-borne CFU-Cs after this lag-time. In contrast, both SEW2871 and FTY720 treatment depleted HSPCs from the lymph within only 6 hours, indicating that S1P, acting primarily through S1P1, controls HSPC exit from non-lymphoid tissues into the draining lymph vessels. This interpretation is also consistent with the observation that prolonged FTY720 treatment resulted in HSPC accumulation in peripheral tissues, while the number of blood-borne HSPCs was reduced. Since HSPCs express S1P1 mRNA and chemotax toward S1P gradients in vitro (suppl. Fig. 4A-C and Fig. 7), it is likely that HSPC-expressed S1P1 mediates HSPC egress from extramedullary tissue. However, it should be noted that lymphatic endothelial cells also express S1P1 (Singer et al., 2005; Wei et al., 2005), which raises the possibility that S1P might exert additional control over HSPC trafficking via its action on lymphatic endothelial cells.
An S1P gradient between lymphoid tissue and lymph fluid is required for lymphocyte egress from LN (Schwab et al., 2005). Interstitial tissue fluids contain low levels of S1P, whereas extracellular S1P concentrations in lymph and plasma are high (Cyster, 2005; Schwab et al., 2005). While plasma S1P is mainly produced by hematopoietic cells, high lymph S1P levels are maintained by a non-hematopoietic, radiation-resistant source (Pappu et al., 2007). The S1P gradient between LNs and lymph fluid is maintained by the S1P-degrading enzyme S1P lyase (Schwab et al., 2005), which is abundant in most tissues and cell types except in platelets and erythrocytes (Pappu et al., 2007). Our present findings suggest that S1P lyase is similarly essential for the egress of HSPCs from non-lymphoid extra-medullary tissues. S1P gradients are also required for the migration of bilateral heart progenitors in zebrafish (Kupperman et al., 2000). Hence, guidance signals provided by extracellular lipids, such as S1P, may represent a conserved navigation mechanism for migrating stem and progenitor cell populations that is utilized by vertebrates and invertebrates alike.
What is the functional relevance of HSPC trafficking in adults? The recirculation of HSPCs between the BM and blood is believed to be important to maintain hematopoietic homeostasis in dispersed BM cavities (Wright et al., 2001b). However, the role of the migratory pool of HSPCs that percolate constitutively through extramedullary tissues and then return to the blood via the TD is less obvious. Our kidney capsule transplantation experiments show that even in the absence of inflammation recirculating HSPCs divide locally in peripheral tissues and differentiate into mature myeloid cells. Homed HSPCs may thus help to constitutively replenish the diverse population of tissue-resident leukocytes that perform multiple essential functions in peripheral organs, including e.g. removal of dead cells and debris.
Given the diverse activities of leukocytes in the context of tissue damage and immune defense, it would seem plausible that HSPCs might possess the ability to sense and respond to situations, such as tissue damage and infections that require the rapid influx of large numbers of innate immune cells. Indeed, HSPCs express TLR2 and TLR4 (Nagai et al., 2006), which sense pathogen-associated molecular patterns, such as peptidoglycan and lipoteichoic acid from Gram-positive bacteria and LPS from Gram-negative bacteria, respectively (Hoshino et al., 1999). It was shown in vitro that incubation of both primitive and committed HSPCs with TLR ligands triggers HSPC proliferation and rapid myeloid differentiation. Based on this discovery, it was hypothesized that this mechanism could provide a means for boosting innate immunity during life-threatening infections, where microbial components either access the circulation and reach the BM or where dispersed stem and progenitor cells sense foreign materials outside the marrow compartment and become locally converted to innate effector cells (Nagai et al., 2006). In fact, we show the existence of a migratory pool of HSPCs that constitutively survey peripheral tissues. Our experiments indicate that once these cells sense microbial danger signals, they proliferate vigorously and boost the local supply of innate effector cells. Thus, the migratory pool of HSPCs might acts as a source of highly versatile stem and progenitor cells that can respond to danger signals locally within tissues before the information has spread to the BM. Notably, TLR stimulation also blocks HSPC egress from inflamed tissues and abolishes HSPC chemotaxis to S1P in vitro. Hence TLR-induced changes in S1P receptor expression regulate the dwell-time of HSPCs in inflamed tissues and thereby act as a rheostat that can boost local leukocyte generation.
In conclusion, we show here that HSPCs constitutively survey extramedullary nonlymphoid tissues. Our results raise the possibility that in the absence of infection migratory HSPCs contribute to the continuous restoration of specialized hematopoietic cells that reside in peripheral tissues. During tissue infection, the migratory pool of HSPCs might act as an immediate and highly adaptive source of progenitor cells that proliferate locally and generate innate immune effector cells. It will be important to determine how HSPC traffic within peripheral tissues is influenced by and may modulate the course of infectious diseases and of other acute or chronic inflammatory conditions.
EXPERIMENTAL PROCEDURES
Thoracic duct cannulation
Thoracic duct cannulation was performed as described previously (Ionac, 2003). Where indicated, mice were treated with PTX (50 μg/kg iv, 2h prior to cannulation), FTY720 (1mg/kg ip, 6h prior to cannulation), SEW2871 (20mg/kg 6h prior to cannulation (Wei et al., 2005)), or THI (drinking water with 10g/L glucose plus 50 μg/ml THI for 3 days prior to cannulation).
Parabiosis
6- to 8-week-old sex- and age-matched C57BL/6 WT and β-actin/ GFP mice (Wright et al., 2001a) were joined at the flanks as described (Wright et al., 2001b). Where indicated, mice were surgically separated prior to collection of TD lymph.
Colony-forming unit assays
MNCs or sorted Lin+, Lin−c-Kit− and Lin−c-Kit+ cells isolated from TD lymph, BM and PB were assessed for CFU-C content by culturing 6 × 106 cells in methylcellulose-containing MethoCult M3434 medium (StemCell Technologies, Vancouver, Canada) as reported (Papayannopoulou et al., 2003).
Primary and secondary transplantation assays
2×107 TD-MNCs collected from CD45.2+C57BL/6 or nude mice were injected into sublethally (900rad) or lethally (1300rad) irradiated CD45.1+ or Rag2−/−γc−/− mice. Where indicated, 105 rescue BM cells from Rag2−/−γc−/− donors were co-injected with TD-MNCs. After 6 weeks, blood was obtained from the retro-orbital plexus and the presence of donor-derived myeloid cells (CD45.2+Gr-1+) or T cells (TCR-β+) was assessed by flow cytometry. For secondary transplantation, 2×107 CD45.2+ TD-MNCs were isolated and transplanted into sublethally irradiated (900 rad) CD45.1+ congenic recipients. Twelve weeks after reconstitution, recipients with pronounced chimerism of peripheral blood granulocytes were sacrificed and donor-derived (CD45.1−) BM cells were sorted using a FACSAria cell sorter (BD Biosciences) and transplanted into lethally irradiated CD45.1+ secondary recipients. Ten weeks later, chimerism among peripheral blood granulocytes, T cells, and B cells (CD19+ B220+) was assessed by FACS analysis.
In vivo assessment of HSPC differentiation
TD-derived GFP+CD45.2+Lin− (5×105), or GFP+CD45.2+Lin−c-Kit+ (5×105), or BM-derived GFP+ Lin−c-Kit+ (5×105), Lin−c-Kit+sca-1-(5×104, or 5×105), or Lin−c-Kit+sca-1+ (5×104) were implanted under the left kidney capsule of CD45.1+ congenic recipient mice. Where indicated, the cells were preincubated with medium containing stem cell factor (R&D, 20 ng/ml), Flt-3 ligand (R&D, 100 ng/ml) in the absence or presence of LPS (Sigma, 10μg/ml) and CD14-Fc (R&D, 1μg/ml) for 12h before implantation or were not pre-exposed to LPS, but injected into the kidneys together with LPS (10μg/ml). The right kidney was used as a control and was injected with medium with or without LPS (Sigma, 10μg/ml) and CD14-Fc (R&D, 1μg/ml). Where indicated, the recipient mice received 1mg/day BrDU i.p (BD Biosciences, San Jose, CA) on days 4, 5 and 6 after implantation. Six days after implantation of the cells, mice were perfusion-fixed and the kidneys were harvested and quantitatively analyzed by confocal laser scanning microscopy. To this end, 5 serial sections (each 30 μm thick) were obtained from the anterior aspect of the kidney and stained using fluorescently tagged anti-GFP (Rockland Inc. Gilbertsville, PA), anti-CD45, anti-CD45.1, anti-CD45.2, anti-Gr-1(Ly6G/C), anti-CD11c, anti-B220, anti-CD3 (all from BD Biosciences), mouse anti-human Ki-67 antibody (B56, cross-reacting with mouse Ki-67, BD Biosciences) and anti-pDC/IPC (clone 120G8.04, Dendritics, France) as indicated. To detect myeloid differentiation, sections were stained with a panel of mAb against myeloid lineage markers (CD11b, CD11c, F4/80, Gr-1(Ly6G/C)), followed by APC-conjugated streptavidin (BD Biosciences). BrDU in situ detection was performed according to the manufacturer's recommendations using biotin-labeled anti-BrDU antibody (BrDU in situ detection kit, BD Biosciences) and APC-conjugated streptavidin (BD Biosciences).
Statistical analyses
Quantitative results are expressed as means±SEM unless otherwise indicated. The Mann–Whitney test was used to determine the significance of differences between means. Statistical significance was assumed at P<0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank G. Cheng, P. Wisniewski, Elisabeth Vollmann, M. Lorenz, S. Kerstan and M. Ionac for their support. Supported by the National Institutes of Health (AI061663, AR42689 and HL56949 to U.H.v.A) and the German Research Foundation DFG (Ma2186/4-1 to S.M.). S.M. is a Heisenberg Scholar of the German Research Foundation DFG. P.S. is supported by SSMBS (Schweizerische Stiftung für Medizinisch-Biologische Stipendien). A.W. is supported by a Burroughs Wellcome Fund Career Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Cardier JE, Barbera-Guillem E. Extramedullary hematopoiesis in the adult mouse liver is associated with specific hepatic sinusoidal endothelial cells. Hepatology. 1997;26:165–175. doi: 10.1002/hep.510260122. [DOI] [PubMed] [Google Scholar]
- Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, Hoshino Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J Immunol. 1998;160:5037–5044. [PubMed] [Google Scholar]
- Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc Natl Acad Sci U S A. 2001;98:14541–14546. doi: 10.1073/pnas.261562798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumano A, Godin I. Ontogeny of the Hematopoietic System. Annu Rev Immunol. 2007;25:745–785. doi: 10.1146/annurev.immunol.25.022106.141538. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, Butcher EC. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming WH, Alpern EJ, Uchida N, Ikuta K, Weissman IL. Steel factor influences the distribution and activity of murine hematopoietic stem cells in vivo. Proc Natl Acad Sci U S A. 1993;90:3760–3764. doi: 10.1073/pnas.90.8.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Goldman JP, Blundell MP, Lopes L, Kinnon C, Di Santo JP, Thrasher AJ. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br J Haematol. 1998;103:335–342. doi: 10.1046/j.1365-2141.1998.00980.x. [DOI] [PubMed] [Google Scholar]
- Goodman JW, Hodgson GS. Evidence for stem cells in the peripheral blood of mice. Blood. 1962;19:702–714. [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Ionac M. One technique, two approaches, and results: thoracic duct cannulation in small laboratory animals. Microsurgery. 2003;23:239–245. doi: 10.1002/micr.10136. [DOI] [PubMed] [Google Scholar]
- Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- Kupperman E, An S, Osborne N, Waldron S, Stainier DY. A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature. 2000;406:192–195. doi: 10.1038/35018092. [DOI] [PubMed] [Google Scholar]
- MacVittie TJ, McCarthy KF. The detection of in vitro monocyte-macrophage colony-forming cells in mouse thymus and lymph nodes. J Cell Physiol. 1977;92:203–207. doi: 10.1002/jcp.1040920208. [DOI] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- Massberg S, von Andrian UH. Fingolimod and sphingosine-1-phosphate--modifiers of lymphocyte migration. N Engl J Med. 2006;355:1088–1091. doi: 10.1056/NEJMp068159. [DOI] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, Engelke K, Xia L, McEver RP, Koni PA, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity. 2005;22:259–270. doi: 10.1016/j.immuni.2005.01.008. [DOI] [PubMed] [Google Scholar]
- McKinney-Freeman SL, Jackson KA, Camargo FD, Ferrari G, Mavilio F, Goodell MA. Muscle-derived hematopoietic stem cells are hematopoietic in origin. Proc Natl Acad Sci U S A. 2002;99:1341–1346. doi: 10.1073/pnas.032438799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Wright DE, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc Natl Acad Sci U S A. 1997;94:1908–1913. doi: 10.1073/pnas.94.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulou T, Priestley GV, Bonig H, Nakamoto B. The role of G-protein signaling in hematopoietic stem/progenitor cell mobilization. Blood. 2003;101:4739–4747. doi: 10.1182/blood-2002-09-2741. [DOI] [PubMed] [Google Scholar]
- Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of Lymphocyte Egress into Blood and Lymph by Distinct Sources of Sphingosine-1-Phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- Singer II, Tian M, Wickham LA, Lin J, Matheravidathu SS, Forrest MJ, Mandala S, Quackenbush EJ. Sphingosine-1-phosphate agonists increase macrophage homing, lymphocyte contacts, and endothelial junctional complex formation in murine lymph nodes. J Immunol. 2005;175:7151–7161. doi: 10.4049/jimmunol.175.11.7151. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- Stein JV, Rot A, Luo Y, Narasimhaswamy M, Nakano H, Gunn MD, Matsuzawa A, Quackenbush EJ, Dorf ME, von Andrian UH. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J Exp Med. 2000;191:61–76. doi: 10.1084/jem.191.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 2002;297:2256–2259. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- Wei SH, Rosen H, Matheu MP, Sanna MG, Wang SK, Jo E, Wong CH, Parker I, Cahalan MD. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6:1228–1235. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DE, Cheshier SH, Wagers AJ, Randall TD, Christensen JL, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 2001a;97:2278–2285. doi: 10.1182/blood.v97.8.2278. [DOI] [PubMed] [Google Scholar]
- Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001b;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.