

Abstract
Formyl peptide receptor (FPR) and C5a receptor (C5aR) are chemoattractant G protein-coupled receptors (GPCRs) involved in the innate immune response against bacterial infections and tissue injury. Like other GPCRs, they recruit β-arrestin1/2 to the plasma membrane and activate the extracellular signal regulated kinases 1 and 2 (ERK1/2). Previous studies with several GPCRs have suggested that β-arrestins play an important role as signal transducers by scaffolding signaling molecules such as ERK1/2. This function of the β-arrestins was not discovered until several years after their role in desensitization and endocytosis had been reported. In this study, we investigated the role of the β-arrestins in the activation of ERK1/2 and receptor endocytosis. We took advantage of previously described mutants of FPR that have defects in Gi coupling or β-arrestin recruitment. The results obtained with the mutant FPRs, as well as experiments using an inhibitor of Gi and cells overexpressing β-arrestin2, showed that activation of ERK1/2 takes place through Gi and is not affected by β-arrestins. However, overexpression of β-arrestin2 does enhance FPR sequestration from the cell surface, suggesting a role in desensitization, as shown for many other GPCRs. Experiments with CHO C5aR cells showed similar sensitivity to the Gi inhibitor as CHO FPR cells, suggesting that the predominant activation of ERK1/2 through G protein may be a common characteristic among chemoattractant receptors.
Keywords: Formyl peptide receptor, Gi, β-arrestin, GPCR, ERK1/2, Cell signaling, C5a receptor, Chemoattractant receptor
Introduction
The mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinases 1 and 2 (ERK1/2), are highly conserved among species ranging from yeast to mammals, and play an important role in cell proliferation and differentiation (reviewed in [1]). For this reason, the various pathways that result in ERK1/2 activation have been extensively studied. Among the most important receptors that activate ERK1/2 are the “seven transmembrane-domain” G protein-coupled receptors (GPCRs). Initially, ERK1/2 activation by GPCRs was thought to be completely dependent on G protein-mediated mechanisms. However, work carried out by Lefkowitz and coworkers elegantly showed a β-arrestin-dependent and G protein-independent pathway: HEK-293 cells transfected with β-arrestin2 siRNA showed almost complete inhibition of ERK1/2 phosphorylation 10–60 min after stimulation of the angiotensin II type 1A receptor (AT1AR) [2]. In contrast, inhibition of the Gs-mediated pathway using a PKC inhibitor reduced ERK1/2 phosphorylation 2 min after stimulation, but had no effect at later time points (30–60 min) when ERK1/2 activation was mediated by β-arrestin. Thus, in addition to the previously recognized roles of β-arrestin as a molecule that arrests the signal by sterically preventing G protein coupling, and that mediates receptor endocytosis through clathrin-coated pits, β-arrestin also plays an important role in ERK1/2 phosphorylation. In addition to the AT1A receptor, β-arrestin-mediated activation of ERK1/2 has been shown with several other GPCRs, such as proteinase-activated receptor 2 [3] and V2 vasopressin receptor [4]. The difference in the timing of G protein- versus β-arrestin-mediated ERK1/2 activation, as described by Ahn and coworkers [2], is likely due to the scaffolding function where β-arrestin coordinates the assembly of the GPCR, ERK1/2 and other signaling molecules on endosomes or the plasma membrane [5]. This temporal difference between β-arrestin- and G protein-mediated ERK1/2 activation presents a useful tool for dissection of the two different pathways.
In addition to a temporal difference, the β-arrestin-mediated pathway also presents a spatial difference: Activation through G protein leads to nuclear translocation of ERK1/2 followed by transcriptional regulation, whereas β-arrestin-mediated activation retains ERK1/2 in the scaffolding complex, preventing nuclear translocation and allowing activation of cytoplasmic targets (reviewed in[1]). The β-arrestin primarily responsible for the scaffolding of MAP kinases has been identified as β-arrestin2 [3,5,6].
An additional level of regulation of ERK1/2 activation has been observed in GPCRs undergoing Gs/Gi switching. These predominantly Gs-coupled receptors switch their coupling to Gi upon phosphorylation of the receptor by protein kinase A (PKA) [7]. The mechanism has been investigated in detail and involves β-arrestin-mediated PDE4 cAMP phosphodiesterase recruitment [8] (see Discussion).
Formyl peptide receptor (FPR) is coupled predominantly to Gi [9–13], and in some cells to G0 and Gz (but not Gs) [14]. We have previously shown that activation of FPR causes rapid and concentration-dependent translocation of β-arrestins from the cytosol to the membrane [15]. To examine what role, if any, β-arrestins play in FPR-mediated activation of ERK1/2, we examined the time course of activation using CHO cells expressing either wild-type FPR, an FPR mutant with a partial defect in Gi coupling (but normal β-arrestin recruitment) (R123A), or a mutant with a defect in β-arrestin recruitment (N297A). We also examined the effect of β-arrestin2 overexpression on ERK1/2 activation and FPR endocytosis. The results from these studies suggested that FPR-mediated ERK1/2 activation is regulated by Gi, whereas β-arrestin2 played a role in receptor sequestration. Additional experiments in NIH-3T3 cells expressing FPR suggested that the result is not specific to CHO cells. Finally, ERK1/2 assays in CHO cells expressing another chemoattractant receptor, C5aR, indicated that chemoattractant receptors other than FPR may also rely on Gi for activation of the ERK1/2 pathway.
Materials and Methods
Cell culture and transfections
FPR mutagenesis, transfection of CHO cells, and characterization of wild-type and mutant FPRs have been described previously [16,17,15]. The FPR haplotype used in this study is haplotype 3 [18] which corresponds to the R-26 cDNA [19] (GenBank accession no M60627). The β-arrestin2 cDNA was amplified by PCR from pOTB7 vector containing the human β-arrestin2 insert (Invitrogen, Inc., Carlsbad, CA) and ligated into the constitutive expression vector pBZSA to allow selection with Bleocin (EMD Biosciences, Inc., La Jolla, CA) [20]. Transfection was carried out using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions; transfectants were selected in 30 μg/ml Bleocin. Two clones with high expression levels of both FPR and β-arrestin2, identified by western blot analysis, were used for all experiments. The CHO C5aR cell line has been characterized previously [21]. CHO cells were maintained in α-modified Eagle’s medium containing 5% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 0.5 mg/ml G418 (cells expressing FPR), and 30 μg/ml Bleocin (cells expressing β-arrestin2). NIH-3T3 cells were transfected with the previously described pBGSA-FPR expression plasmid and clones were selected by immunofluorescence microscopy [22]. Increased expression was induced by adding 6 mM sodium butyrate to the medium 14 to 16 hours before each experiment [23].
Western blot analysis
The β-arrestin membrane translocation assay and the ERK1/2 activation assay have been previously described [15]. Wild-type and mutant FPR were detected with monoclonal antibody NFPR1, and phosphorylated FPR was identified by loss of NFPR2 monoclonal antibody binding to ligand-activated receptor [24]. In Gi protein inhibition experiments, cells were incubated 14–16 h in the presence of 100 ng/ml pertussis toxin (Sigma, Inc., St. Louis, MO).
Immunofluorescence endocytosis assay
Cells were incubated in the absence or presence of 1 μM fMLF for the indicated times, fixed in 2.5% paraformaldehyde/PBS, permeabilized with 0.01% saponin/PBS and FPR was labeled with a polyclonal antibody against the C-terminal 12 amino acids of FPR and Alexa™-488-conjugated secondary antibody (Molecular Probes). Images were acquired using a Zeiss Axioscope 2-Plus microscope and imaging system (Carl Zeiss Microimaging, Inc., Thornwood, NY).
Flow cytometer endocytosis assay
Cell were incubated at 37ºC for 0, 5, 10, 20 or 60 min with 100 nM fMLF, washed with PBS, pH 3.0, followed by PBS, pH 7.4. PBS containing 20 nM formyl-Norleucine-Lucine-Phenylalanie-Norleucine-Tyrosine-Lysine-fluorescein (Molecular Probes, Inc., Eugene, OR) was added to the cells and the cells were immediately scraped off the plate. Half of the volume was placed in empty flow cytometer tubes and the other half was placed in tubes containing 20 μM fMLF to detect background binding, which was subtracted. The cells were kept on ice for 45 min prior to flow cytometry. The fluorescence of 10,000 cells was measured and the amount of FPR on the cell surface was calculated as the % of the fluorescence obtained in the absence of ligand (100%). Experiments were carried out in triplicate.
Results
Inhibition of Gi with pertussis toxin blocks FPR-mediated activation of ERK1/2
It has been previously shown that receptors coupled to Gs, such as angiotensin II type 1A receptor (AT1AR) and type 1 parathyroid hormone (PTH)/PTH-related peptide receptor (PTH1R) induce rapid, but transient, activation of ERK1/2 through G protein (maximal activation at 2 and 10 min, respectively), whereas ERK1/2 activation through β-arrestin is delayed and prolonged (maximal at 10–60 min) [2,25]. To examine the role of Gi and β-arrestin1/2 in FPR-mediated activation of ERK1/2, we analyzed the effect of pertussis toxin, an inhibitor of Gi, on phosphorylation of ERK1/2 over a time course of 0–30 min. Our hypothesis was that since pertussis toxin blocks activation through Gi, we would not see activation of ERK1/2 at 2–10 min after addition of ligand. However, if β-arrestin1/2 (that is not blocked by pertussis toxin) mediates activation of ERK1/2, then we would detect phosphorylated ERK1/2 at 20–30 min after addition of ligand. Figure 1A shows that incubation with pertussis toxin completely inhibited ERK1/2 phosphorylation, not only at 2, 5 and 10 min, but also at 20 and 30 min, suggesting that β-arrestin1/2 may not play a role in FPR-mediated activation of ERK1/2 (Figure 1A). To rule out the possibility that pertussis toxin inhibited receptor phosphorylation and/or β-arrestin1/2 recruitment, thereby preventing β-arrestin1/2–mediated activation of ERK1/2, we examined receptor phosphorylation and β-arrestin1/2 recruitment in the absence and presence of pertussis toxin. We took advantage of a recently characterized monoclonal antibody against FPR that does not bind to phosphorylated receptor [24]. Western blot analysis of membrane fractions from CHO FPR cells incubated for 8 min with various concentrations of ligand (0, 0.1, 1, 10 μM fMLF) showed no difference in receptor phosphorylation between cells incubated in the absence or presence of pertussis toxin (Figure 1B). Similarly, pertussis toxin did not affect ligand-induced recruitment of β-arrestin1/2 from the cytosol to the membrane (Figure 1C). Therefore, the observed block in FPR-mediated ERK1/2 activation in the presence of pertussis toxin was not due to a harmful effect on β-arrestin1/2.
Figure 1. Pertussis toxin inhibits FPR-mediated activation of ERK1/2, but does not affect phosphorylation of FPR or FPR-induced recruitment of β-arrestin.

Cells were incubated overnight in the absence or presence of 100 ng/ml pertussis toxin prior to activation with fMLF. (A) Cells were exposed to 100 nM fMLF for 0–30 min. The relative amounts of phosphorylated ERK1/2 (upper panel) and total ERK1/2 (lower panel) were examined by western blotting. (B) Cells were incubated for 8 min with various concentrations of fMLF and the phosphorylation of FPR was visualized by western blotting using an anti-FPR monoclonal antibody (NFPR2) that does not bind phosphorylated receptor. (C) Cells were activated for 8 min with the indicated concentrations of fMLF. β-arrestins in the membrane (m) and cytosol (c) fractions were detected by western blotting.
Characterization of wild-type and mutant FPRs
Since pertussis toxin could have unknown adverse effects on other cell signaling molecules, we took advantage of two previously described FPR mutants to further examine the role of Gi and β-arrestin in FPR-mediated activation of ERK1/2: FPR N297A has a defect in phosphorylation and membrane translocation of β-arrestin1/2, and FPR R123A has a partial defect in G protein coupling, but normal β-arrestin1/2 translocation [16,22]. Figure 2A shows the differences in ligand-induced membrane translocation of β-arrestin1/2 [15], and Figure 2B shows the defect in ligand-induced phosphorylation of FPR N297A [17]. These results confirm our previous findings and allow us to use these mutants as tools to examine ERK1/2 activation.
Figure 2. CHO FPR N297A is defective in β-arrestin recruitment and receptor phosphorylation.
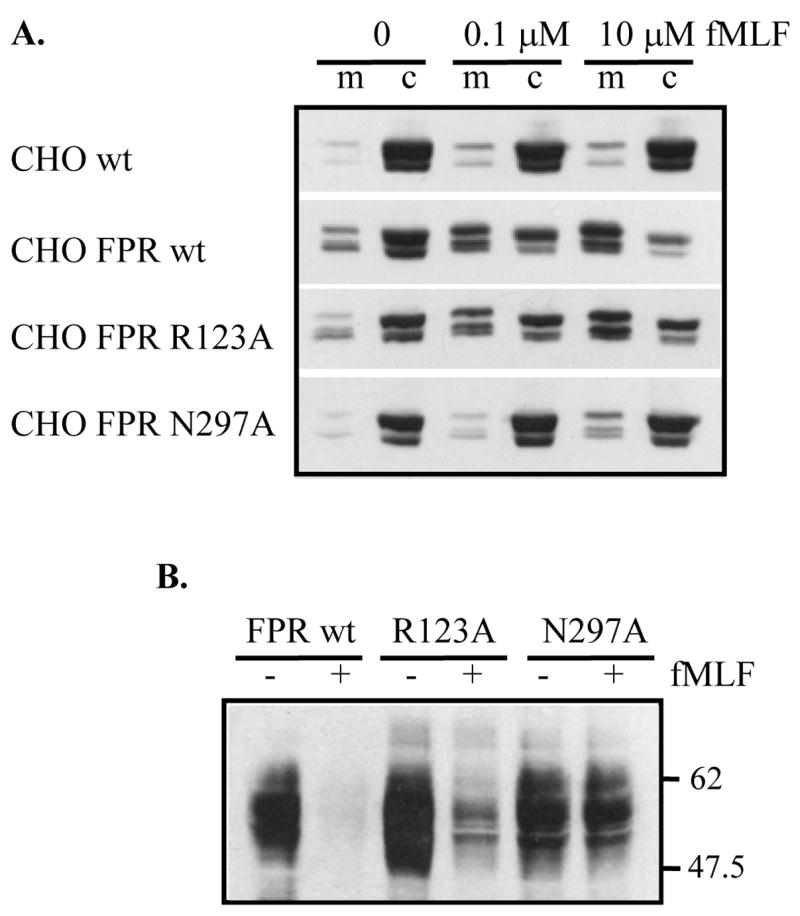
(A) Cells were incubated for 8 min in the absence or presence of fMLF, as shown. β-arrestin was detected in membrane (m) and cytosol (c) fractions by western blotting. CHO wild type cells that do not express FPR were used as a negative control. (B) Cells were incubated for 8 min with (+) or without (−) 10 μM fMLF. Phosphorylated FPR was identified as loss of NFPR2 antibody binding in a western blot.
Wild-type FPR, FPR R123A and FPR N297A show identical time courses of ERK1/2 activation
Our hypothesis, based on the results presented in Figure 1 (suggesting that ERK1/2 is regulated mainly by Gi), was that CHO FPR N297A cells would show a similar decline in ERK1/2 activation as seen in CHO wt FPR cells. On the other hand, a more rapid decline in CHO FPR N297A cells compared to CHO FPR wt cells would suggest that the ERK1/2 phosphorylation at later time points (20–30 min) in CHO FPR wt cells indeed is mediated by β-arrestin. In contrast, our other FPR mutant, FPR R123A, with a partial defect in Gi coupling, would show Gi-mediated ERK1/2 activation only at high concentrations of ligand, unless the ERK1/2 activation is mediated by β-arrestin. The cells were incubated with a saturating concentration (10 μM) of fMLF for 0, 1, 2, 4, 8, 16 or 32 minutes. Representative western blots of the time courses of ERK1/2 activation by wild-type FPR, FPR R123A and FPR N297A are shown in Figure 3A, and the quantification from three separate experiments is presented in Figure 3B. As shown, FPR R123A exhibits a similar time course of ERK1/2 activation compared to wild-type receptor. At nanomolar concentrations of ligand, the amount of phosphorylated ERK1/2 is decreased, but the time course remains identical (data not shown). Conversely, FPR N297A displays a prolonged activation with a statistically higher level of phosphorylated ERK1/2 at 16 min (P = 0.004) and 32 min (P = 0.036). This would suggest that a β-arrestin1/2-mediated pathway is involved. However, we know from our previous characterization of FPR N297A that this mutant does not recruit β-arrestin1/2 and does not become desensitized to ligand [17]. Thus, the prolonged activation is more likely due to lack of receptor desensitization. To explore this further, we examined whether incubation with pertussis toxin will inhibit the ERK1/2 activation at 16 and 32 min. Figure 3C shows essentially complete inhibition of ERK1/2 phosphorylation in the presence of the toxin, indicating that the activation at later time points was indeed mediated by Gi rather than β-arrestin1/2.
Figure 3. Time course analysis in combination with pertussis toxin treatment does not support a role for β-arrestins in ERK1/2 phosphorylation.
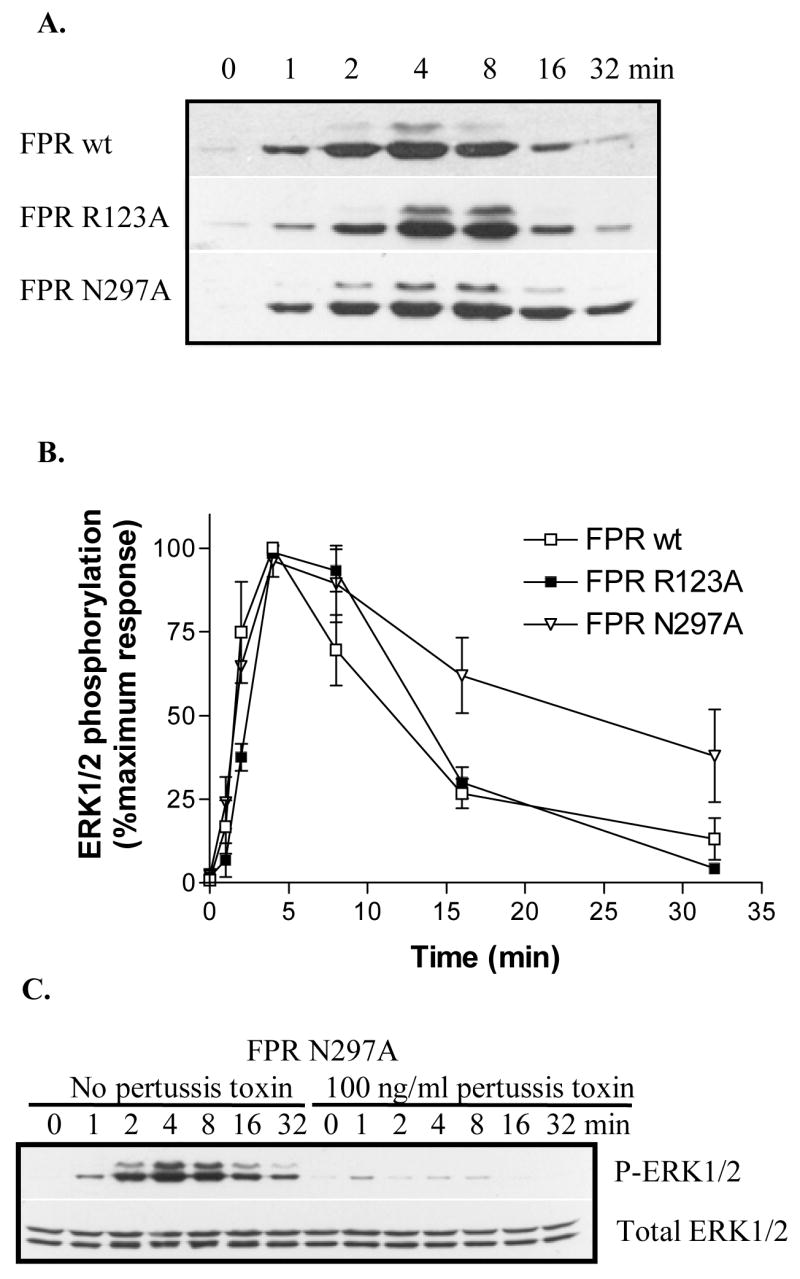
(A) CHO cells expressing wild-type FPR, FPR R123A mutant, or FPR N297A mutant were exposed to 10 μM fMLF for 0, 1, 2, 4, 8, 16 or 32 min. Phosphorylated ERK1/2 in whole cell extract was detected by western blotting. (B) The relative amount of phosphorylated ERK1/2 was quantified by scanning individual western blots. The results are means ± SD from a minimum of three different experiments for each receptor. (C) CHO FPR N297A cells were incubated overnight in the absence or presence of 100 ng/ml pertussis toxin to inhibit Gi. Cells were exposed to 10 μM fMLF for 0, 1, 2, 4, 8, 16 or 32 min. Phosphorylated ERK1/2 (upper panel) and total ERK1/2 (lower panel) from whole cell extracts were detected by western blotting.
Overexpression of β-arrestin2 does not amplify ERK1/2 activation
To address the possibility that the expression level of β-arrestin2 (the β-arrestin mainly responsible for ERK1/2 scaffolding) is not expressed at sufficient levels to result in visible activation of ERK1/2 in CHO cells, we transfected β-arrestin2 into cells already expressing wild-type FPR. The expression levels for FPR, β-arrestin1/2 and ERK1/2 were examined by western analysis. Note the approximately 3-fold higher expression level of β-arrestin2 in the double transfectants (Figure 4A). First, we analyzed whether the increased amount of β-arrestin2 would affect the concentration dependence of ERK1/2 activation at 8 min of stimulation. No significant difference could be detected compared to cells expressing endogenous β-arrestin1/2 only (Figure 4B and C). Next, we examined the time course of ERK1/2 activation, as previously shown in Figure 3. Again, the cells overexpressing β-arrestin2, showed a time course essentially identical to that of CHO FPR cells, suggesting that the increased amount of β-arrestin2 did not amplify ERK1/2 phosphorylation (Figure 5 A and B).
Figure 4. Overexpression of β-arrestin2 does not affect the concentration dependence of ERK1/2 activation.

(A) Expression levels of FPR, β-arrestin1/2 and ERK1/2 were examined in CHO FPR cells (1) and CHO FPR + β-arrestin2 cells (2) by western blot. (B) The concentration dependence of ERK1/2 activation was examined by incubation of CHO FPR cells and CHO FPR + β-arrestin2 cells for 4 min with various concentrations of fMLF, as shown. Phosphorylated ERK1/2 was identified by western blotting. (C) The relative amount of phosphorylated ERK1/2 was quantified by scanning western blots from three different experiments and show means ± SD. The differences between the two cell lines were not statistically significant.
Figure 5. Overexpression of β-arrestin2 does not affect the time course of ERK1/2 activation.
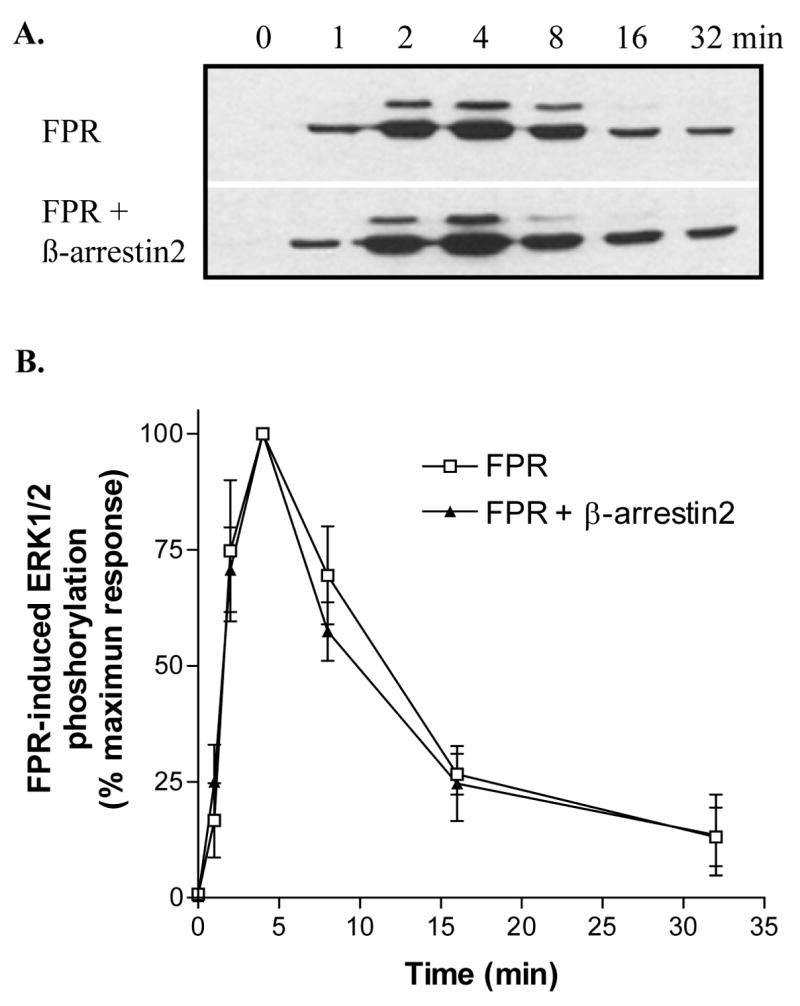
(A) Cells were incubated for the indicated times with 100 nM fMLF and phosphorylated ERK1/2 was visualized in western blots. (B) FPR-induced ERK1/2 phosphorylation was quantified from three western blots by scanning. Results show means ± SD.
Overexpression of β-arrestin2 causes a time-dependent increase in the amount of internalized FPR
Since it is also known that β-arrestin1/2 mediates GPCR endocytosis through clathrin-coated pits, we examined whether overexpression of β-arrestin2 enhances internalization of FPR in CHO cells. Immunofluorescence staining of FPR after incubation with ligand showed a clear difference in the localization of FPR in the two different cell lines. As seen in Figure 6A, cells overexpressing β-arrestin2 had a more punctate staining, suggesting more efficient endocytosis. To obtain more quantitative data, we carried out flow cytometer analysis of the amount of receptor on the cell surface at different times of ligand incubation (Figure 6B). These experiments clearly indicated that overexpression of β-arrestin2 resulted in an increased amount of intracellular FPR, compared to cells containing only endogenous β-arrestin1/2. Since these experiments do not distinguish between the possibility that overexpression results in more efficient endocytosis and/or less efficient recycling to the cell surface, we can only conclude that β-arrestin2 increases the amount of internalized FPR.
Figure 6. Overexpression of β-arrestin2 enhances ligand-induced sequestration of FPR from the cell surface.
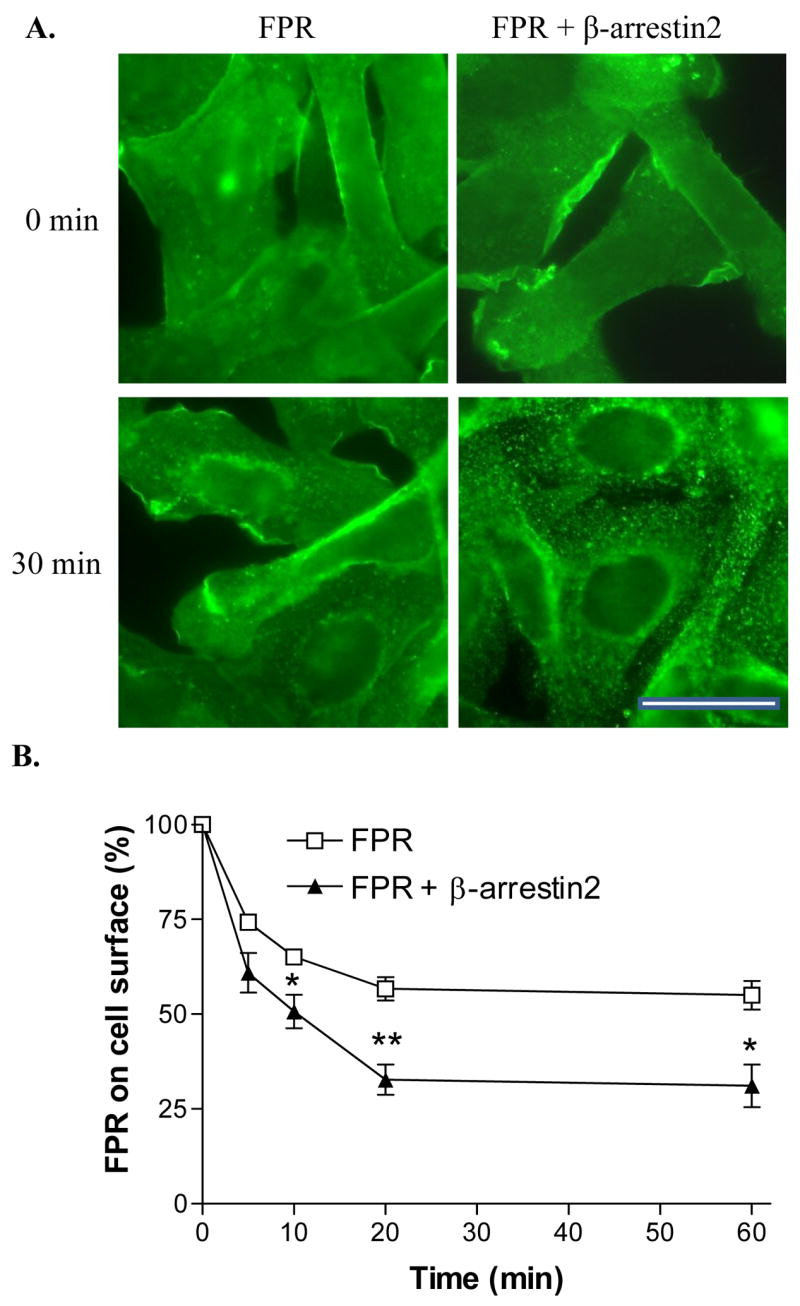
(A) CHO FPR and CHO FPR + β-arrestin2 cells were incubated for 0 or 30 min in the presence of 1 μM fMLF and subsequently stained with a polyclonal anti-FPR antibody. Bar 50 μm. (B) CHO FPR and CHO FPR + β-arrestin2 cells were incubated for 0, 5, 10, 20 or 60 min in the presence of 100 nM fMLF, washed in low pH buffer and the receptors remaining on the cell surface were detected by flow cytometry after 45 min binding of fluorescent ligand on ice. Graph shows means ± SD from three separate experiments. * P < 0.05; **P < 0.01.
FPR-mediated activation of ERK1/2 in NIH-3T3 transfectants is similar to that in CHO transfectants
To examine the possibility that our results are cell type dependent, we decided to analyze ERK1/2 activation by FPR in another cell line. NIH-3T3 cells were transfected with FPR and a time course experiment in the absence and presence of pertussis toxin was carried out (Figure 7A and C). The increase in ERK1/2 phosphorylation showed a similar time dependence in NIH-3T3 transfectants compared to CHO transfectants with maximal activation at 4 min, although the activation curve was even more transient in NIH-3T3 transfectants than in CHO transfectants with statistically significant differences at 2 min (P = 0.015) and 16 min (P = 0.003). The results upon treatment with pertussis toxin confirmed that the ERK1/2 activation is mediated predominantly by G protein (Figure 7A and C). Thus the results seen with the CHO FPR cells do not appear to be specific for that particular cell line.
Figure 7. ERK1/2 activation is blocked by pertussis toxin in NIH-3T3 cells expressing FPR and in CHO cells expressing C5aR.
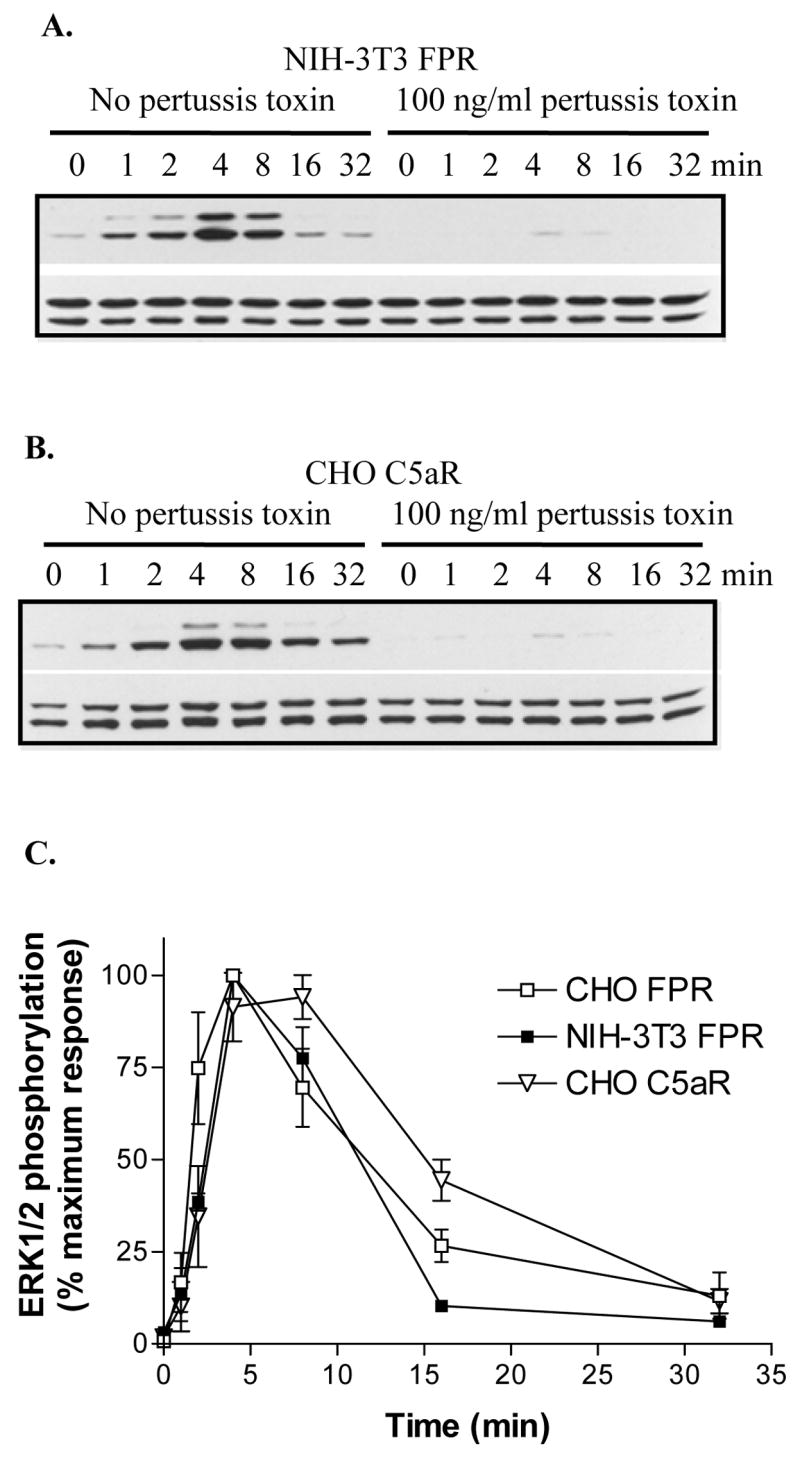
(A) NIH-3T3 FPR cells were incubated overnight in the absence or presence of 100 ng/ml pertussis toxin. Cells were activated for the indicated times with 10 μM fMLF and phosphorylated ERK1/2 (upper panel) and total ERK1/2 (lower panel) were detected from whole cell extracts by western blot analysis. (B) The effect of pertussis toxin on C5aR-induced ERK1/2 activation was examined as above. (C) The relative amount of phosphorylated ERK1/2 was quantified from western blots by scanning. The results show means ± SD from three different experiments.
Another chemoattractant receptor, the complement fragment C5a receptor, also activates ERK1/2 primarily through G protein
To consider the possibility that the GPCRs that belong to the chemoattractant receptor family activate ERK1/2 primarily through Gi and not through β-arrestin, we also analyzed the time course of ERK1/2 activation through C5aR in CHO transfectants in the absence and presence of pertussis toxin. The time course for C5aR-mediated ERK1/2 phosphorylation was similar to that seen in FPR-expressing cells, although interestingly the activation of ERK1/2 was somewhat delayed and prolonged compared to that by FPR with statistically different results observed at 2 min (P = 0.014), 8 min (P = 0.011), and 16 min (P = 0.004). The prolonged activation could theoretically be due to β-arrestin1/2–mediated phosphorylation of ERK1/2. However, almost complete inhibition with pertussis toxin was observed, ruling out a role for β-arrestin1/2 in ERK1/2 activation (Figure 7B). We also observed that overexpression of β-arrestin2 increases the internalized pool of C5aR upon incubation with C5a, in a fashion similar to that seen with CHO cells expressing FPR and β-arrestin2 (data not shown). This suggests that C5aR, and possibly other chemoattractant receptors, activate ERK1/2 mainly through Gi with minimal or no contribution by β-arrestin1/2, whereas β-arrestin1/2 participates in receptor desensitization and endocytosis.
Discussion
Diverse ligands including neurotransmitters, hormones, growth factors and chemokines bind GPCRs, resulting in phosphorylation of ERK1/2 and activation of specific cellular targets in the cytoplasm and/or the nucleus. It is therefore no surprise that the regulation of the ERK1/2 signaling cascade by GPCRs is highly complex and cell type specific [7]. For example, PKA-mediated phosphorylation of the β1 and β2 adrenergic receptors switches the coupling from Gαs to Gαi resulting in pertussis toxin sensitive activation of ERK1/2 [26,27]. The underlying mechanism of the switch was described by Houslay and co-workers who showed that a β-arrestin-sequestered subpopulation of the PDE4D5 isoform specifically regulated the phosphorylation of β2 adrenergic receptor by PKA (reviewed in [28,29]). Thus, the activation of ERK1/2 by GPCRs coupled to Gs/Gi is highly complex and regulated on multiple levels by a large number of signaling molecules.
The role of β-arrestin in ERK1/2 activation by Gs-coupled GPCRs has been extensively documented [30]. However, few studies have been carried out using Gi-coupled GPCRs (that do not couple to Gs). We examined the role of β-arrestin in FPR-mediated activation of ERK1/2 using two different approaches 1) inhibition of Gi with pertussis toxin, and 2) analysis of mutant FPRs with β-arrestin-binding or Gi-coupling defects.
Previous experiments showed that pertussis toxin inhibits FPR-mediated activation of ERK1/2, suggesting that activation is dependent on Gi [31,32]. However, since these studies were carried out 0–5 min post stimulation, and other GPCRs have been shown to induce maximal β-arrestin-mediated ERK1/2 signaling at 10–30 min post stimulation [2,25], we sought to establish whether the effect previously detected with pertussis toxin would also apply at later times points of stimulation. Our results with both CHO and NIH-3T3 FPR transfectants clearly showed that pertussis toxin inhibited ERK1/2 phosphorylation both at early time points (1–4 min) and at later time points (8–32 min).
Mutant GPCRs that couple to G protein, but do not recruit β-arrestins, or mutants that do not couple to G proteins, but still recruit β-arrestins in response to ligand stimulation, provide excellent models to study the contributions of G proteins and β-arrestins in ERK1/2 activation. To date, only one study with this type of mutation has been reported to our knowledge: A DRY/AAY mutation in the angiotensin type 1A receptor (AT1AR) uncouples the receptor from Gs while retaining the capacity to recruit β-arrestins and activate ERK1/2 [33]. The R in the DRY motif of AT1AR corresponds to R123 in FPR. The R is also found in most other GPCRs and it affects G protein coupling/recognition through its role in regulating the receptors’ conformational state (for review, see [34]). Our previous analysis of FPR R123A has shown that its signaling through Gi is severely impaired, but not abolished [15,16]. While wild-type FPR mediated maximal ERK1/2 activation at ~5 nM fMLF, FPR R123A required ~1 μM fMLF for maximal activation, a 200-fold difference in concentration. Similarly, the EC50 for fMLF-induced intracellular calcium release by FPR R123A was about 250-fold higher than that of wild-type FPR [15]. Another mutation, N297A, in the highly conserved NPXXY motif found in the C-terminal end of the 7th transmembrane domain of most GPCRs also turned out to be useful in our analysis of β-arrestin-mediated activation of ERK1/2. We have earlier shown that this mutation prevents ligand-induced phosphorylation of FPR and recruitment of β-arrestin1/2 [15,17]. Thus, a time course analysis similar to that previously carried out for AT1AR seemed likely to reveal the contribution of Gi and β-arrestin1/2 in ERK1/2 activation by FPR. Indeed, comparing these two mutants with wild-type FPR, we concluded that the phosphorylation of ERK1/2 upon receptor activation is primarily regulated by Gi.
β-Arrestins’ role as endocytic adapters linking GPCRs to clathrin-coated pits has been well established (for review, see [35]). It has also been shown that overexpression of wild-type β-arrestins enhanced endocytosis of β2-adrenergic receptor, whereas mutant β-arrestins inhibited endocytosis [36]. It was therefore no surprise that overexpression of β-arrestin2 in CHO transfectants enhanced the sequestration of FPR. This result also confirms that the expression level of β-arrestin2 was sufficient to observe a functional difference between the cell lines, further validating the result of the ERK1/2 experiment where no shift in the time course of ERK1/2 phosphorylation could be observed despite the overexpression of β-arrestin2.
Our data suggest that both FPR and C5aR activate ERK1/2 primarily through Gi. This may be a common feature for the classical chemoattractant receptors. However, it does not appear to apply to chemokine receptors, such as CCR7 since a study by Kohout and coworkers showed partial inhibition of CCL19-induced ERK1/2 activation in cells treated with β-arrestin2 siRNA [37]. In a recent study by Huet and coworkers, the role of β-arrestins in the formyl peptide receptor-like-1 (FPRL1) internalization and signaling was examined [38]. In a mouse embryonic fibroblast (MEF) cell line lacking endogenous β-arrestins, the internalization was highly compromised, whereas ERK1/2 phosphorylation was rapid and transient and could be inhibited with pertussis toxin. Thus, like FPR, FPRL1 mediated ERK1/2 activation primarily through Gi [38]. However, the reduced FPRL1 internalization result differed from that obtained with FPR in MEF β-arrestin1/2 knock-out cells, where the absence of both β-arrestins did not affect endocytosis [39]. In addition, cells expressing dominant negative β-arrestins or partially phosphorylated mutant FPRs that do not bind wild-type β-arrestin, have been shown to internalize FPR at normal rate and extent [40,41]. Thus, β-arrestins are not required for internalization of FPR; however, based on our results with cells overexpressing β-arrestin2, an increased amount of β-arrestin does enhance sequestration. Since a previous study using the MEF β-arrestin knockout cells has shown that β-arrestins are required for recycling of FPR, our result showing an increasing amount of intracellular FPR in cells overexpressing β-arrestin2 is presumably due to enhanced endocytosis, rather than reduced recycling [39].
We have previously shown that FPR-mediated activation of ERK1/2 results in phospho-ERK1/2 translocation to the nucleus [15]. Since scaffolding of ERK1/2 by β-arrestins retains activated ERK1/2 in the cytosol and prevents nuclear entry and transcriptional activation, we propose that the lack of β-arrestin scaffolding of ERK1/2 upon activation by FPR may ensure that the innate immune response is fully activated, including the transcriptional regulation and synthesis of cytokines [42].
Acknowledgments
We thank Elena Suvorova and Bruce Granger for advice, Robert Lefkowitz (Howard Hughes Medical Institute and Duke University Medical Center, Durham, NC) for antibody against β-arrestins, and Jim Burritt and Al Jesaitis for equipment usage. This work was supported by National Institutes of Health Grant AI51726 (to H.M.M.) and American Heart Association Grant-in-Aid 060153Z (to H.M.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Werry TD, Christopoulos A, Sexton PM. Curr Pharm Des. 2006;12:1683. doi: 10.2174/138161206776873725. [DOI] [PubMed] [Google Scholar]
- 2.Ahn SK, Shenoy SK, Wei HJ, Lefkowitz RJ. J Biol Chem. 2004;279:35518. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 3.DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. J Cell Biol. 2000;148:1267. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charest PG, Bouvier M. J Biol Chem. 2003;278:41541. doi: 10.1074/jbc.M306589200. [DOI] [PubMed] [Google Scholar]
- 5.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc Natl Acad Sci USA. 2001;98:2449. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Science. 2000;290:1574. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 7.Lefkowitz RJ, Pierce KL, Luttrell LM. Mol Pharmacol. 2002;62:971. doi: 10.1124/mol.62.5.971. [DOI] [PubMed] [Google Scholar]
- 8.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. Proc Natl Acad Sci USA. 2003;100:940. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Bokoch GM. Blood. 1995;86:1649. [PubMed] [Google Scholar]
- 10.Klinker JF, Wenzel-Seifert K, Seifert R. Gen Pharmacol. 1996;27:33. doi: 10.1016/0306-3623(95)00107-7. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki T, Hazeki O, Hazeki K, Ui M, Katada T. Biochim Biophys Acta. 1996;1313:72. doi: 10.1016/0167-4889(96)00048-1. [DOI] [PubMed] [Google Scholar]
- 12.Alemany R, Heringdorf DMZ, Van Koppen CJ, Jakobs KH. J Biol Chem. 1999;274:3994. doi: 10.1074/jbc.274.7.3994. [DOI] [PubMed] [Google Scholar]
- 13.Bennett TA, Key TA, Gurevich VV, Neubig R, Prossnitz ER, Sklar LA. J Biol Chem. 2001;276:22453. doi: 10.1074/jbc.M009679200. [DOI] [PubMed] [Google Scholar]
- 14.Tsu RC, Lai HWL, Allen RA, Wong YH. Biochem J. 1995;309:331. doi: 10.1042/bj3090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gripentrog JM, Miettinen HM. Cell Signal. 2005;17:1300. doi: 10.1016/j.cellsig.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Miettinen HM, Gripentrog JM, Mason MM, Jesaitis AJ. J Biol Chem. 1999;274:27934. doi: 10.1074/jbc.274.39.27934. [DOI] [PubMed] [Google Scholar]
- 17.Gripentrog JM, Jesaitis AJ, Miettinen HM. Biochem J. 2000;352:399. [PMC free article] [PubMed] [Google Scholar]
- 18.Sahagun-Ruiz A, Colla JS, Juhn J, Gao JL, Murphy PM, McDermott DH. Genes Immun. 2001;2:335. doi: 10.1038/sj.gene.6363787. [DOI] [PubMed] [Google Scholar]
- 19.Boulay F, Tardif M, Brouchon L, Vignais P. Biochemistry. 1990;29:11123. doi: 10.1021/bi00502a016. [DOI] [PubMed] [Google Scholar]
- 20.Gripentrog JM, Kantele KP, Jesaitis AJ, Miettinen HM. J Immunol. 2003;171:3187. doi: 10.4049/jimmunol.171.6.3187. [DOI] [PubMed] [Google Scholar]
- 21.Suvorova ES, Gripentrog JM, Miettinen HM. Traffic. 2005;6:100. doi: 10.1111/j.1600-0854.2004.00256.x. [DOI] [PubMed] [Google Scholar]
- 22.Miettinen HM, Mills JS, Gripentrog JM, Dratz EA, Granger BL, Jesaitis AJ. J Immunol. 1997;159:4045. [PubMed] [Google Scholar]
- 23.Gorman CM, Howard BH, Reeves R. Nucl Acids Res. 1983;11:7631. doi: 10.1093/nar/11.21.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riesselman M, Miettinen HM, Gripentrog JM, Lord CI, Mumey B, Dratz EA, Taylor RM, Jesaitis AJ. J Immunol. 2007;179:2520. doi: 10.4049/jimmunol.179.4.2520. [DOI] [PubMed] [Google Scholar]
- 25.Gesty-Palmer D, Chen MY, Reiter E, Ahn S, Nelson CD, Wang ST, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. J Biol Chem. 2006;281:10856. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 26.Daaka Y, Luttrell LM, Lefkowitz RJ. Nature. 1997;390:88. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 27.Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. Cell Signal. 2004;16:1397. doi: 10.1016/j.cellsig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Houslay MD, Baillie GS, Maurice DH. Circ Res. 2007;100:950. doi: 10.1161/01.RES.0000261934.56938.38. [DOI] [PubMed] [Google Scholar]
- 29.Lynch MJ, Baillie GS, Houslay MD. Biochem Soc Trans. 2007;35:938. doi: 10.1042/BST0350938. [DOI] [PubMed] [Google Scholar]
- 30.Dewire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Annu Rev Physiol. 2007;69:483. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 31.Torres M, Ye RD. J Biol Chem. 1996;271:13244. doi: 10.1074/jbc.271.22.13244. [DOI] [PubMed] [Google Scholar]
- 32.Rane MJ, Carrithers SL, Arthur JM, Klein JB, McLeish KR. J Immunol. 1997;159:5070. [PubMed] [Google Scholar]
- 33.Wei HJ, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Proc Natl Acad Sci USA. 2003;100:10782. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rovati GE, Capra V, Neubig RR. Mol Pharmacol. 2007;71:959. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 35.Moore CA, Milano SK, Benovic JL. Annu Rev Physiol. 2007;69:451. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 36.Ferguson SSG, Downey WEI, Colapietro AM, Barak LS, Mènard L, Caron MG. Science. 1996;271:363. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 37.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. J Biol Chem. 2004;279:23214. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 38.Huet E, Boulay F, Barral S, Rabiet MJ. Cell Signal. 2007;19:1939. doi: 10.1016/j.cellsig.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Vines CM, Revankar CM, Maestas DC, LaRusch LL, Cimino DF, Kohout TA, Lefkowitz RJ, Prossnitz ER. J Biol Chem. 2003;278:41581. doi: 10.1074/jbc.C300291200. [DOI] [PubMed] [Google Scholar]
- 40.Gilbert TL, Bennett TA, Maestas DC, Cimino DF, Prossnitz ER. Biochemistry. 2001;40:3467. doi: 10.1021/bi001320y. [DOI] [PubMed] [Google Scholar]
- 41.Bennett TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. J Biol Chem. 2001;276:49195. doi: 10.1074/jbc.M106414200. [DOI] [PubMed] [Google Scholar]
- 42.Cassatella MA, Bazzoni F, Ceska M, Ferro I, Baggiolini M, Berton G. J Immunol. 1992;148:3216. [PubMed] [Google Scholar]