

Introduction
Amyloidosis is a group of rare diseases in which a specific protein is deposited as aggregated interstitial fibrils that can compromise organ function and lead to death.1 In the case of immunoglobulin light chain amyloidosis (AL), the specific protein is the light chain of the monoclonal gammopathy of a plasma cell dyscrasia. This is a rare disorder occurring at an incidence of 9 per million per year,2 which is about 1/5 common as multiple myeloma.
The mechanism by which AL amyloid fibrils are formed is not well understood. It is postulated that the immunoglobulins are secreted, most commonly with an excess of unchaparoned free light chains.3,4 These light chains are then somehow proteolyzed and/or processed in to oligomers and finally non-branching fibrils (8-10 nm) that are deposited in the microcirculation. In the case of renal involvement, it has been postulated that some of this process occurs in mesangial cells.5
Presenting Symptoms and Signs
The spectrum of presentations is diverse, since this is a systemic disease that can affect almost any organ system outside of the central nervous system. A high index of suspicion is required for prompt diagnosis and avoidance of unnecessary morbidity and mortality. The two most frequently involved organs systems are the kidney and the heart, followed by the liver and nervous system. 2,6,7Figure 1a demonstrates the dominant organ presentation during a 10 year period at two amyloid treatment centers. Pulmonary, lymph node, and muscular involvement is also possible, but is less common and more difficult to document in the case of pulmonary and muscle.
Figure 1.
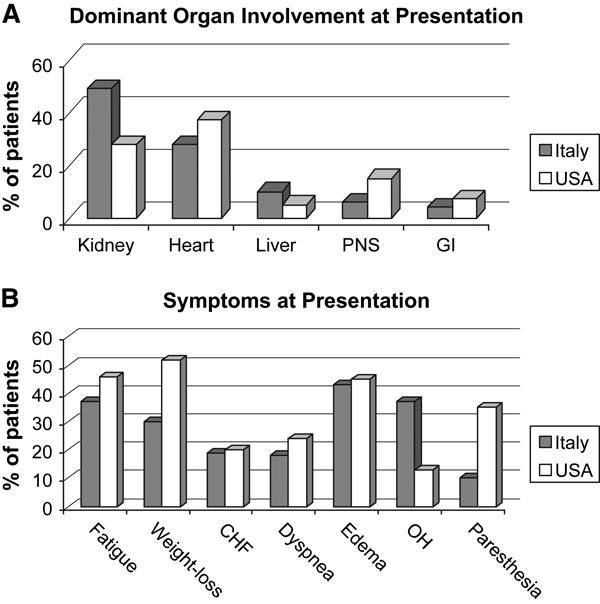
Presenting amyloid syndromes and symptoms at two large amyloid centers
A. Dominant organ involvement at presentation.
B. Symptoms at presentation.
Based on data from: Palladini G et al. Amyloid. 2005;12:120-126
Classic symptoms and signs such as dramatic periorbital purpura, macroglossia, and the shoulder pad sign are pathogno0monic, but each occurs in fewer than 10-15% of patients, making them inadequate to make a timely diagnosis of the syndrome for most patients.
One of the most common presenting symptoms is peripheral edema (Figure 1b), which occurs in just over 40% of patients. The most common cause for edema is hypoalbuminemia due to nephrotic syndrome, but it can also be due to heart failure or obstructive liver disease.
Fatigue is present in 35 to 45% of patients, while dyspnea is present in approximately 20%. These symptoms often represent cardiac involvement, which may go undiagnosed due to unrecognized subtle diastolic dysfunction and mild to moderate left ventricular hypertrophy, because of the preservation of left ventricular ejection fraction.
Orthostatic hypotension is another common finding. This too can be due to one or more of the following: volume contraction from overzealous diuretic therapy to treat nephrotic syndrome or heart failure, autonomic nervous system involvement, or low cardiac output.
Ten to 35% of patients may report paresthesias. The paresthesias in the hands are most commonly due to median nerve entrapment by build-up of amyloid at the carpal ligament. The paresthesias in the feet tend to be dysesthetic in quality and due to a small fiber peripheral neuropathy.
Symptomatic gastrointestinal involvement is rare, but biopsies are positive in the majority of patients sampled. The reason for this disparity relates to the principle that amyloid affects microvessels and may therefore be found in virtually any biopsy sample with a vessel. With the exception of claudication-type symptoms, as a rule an organ becomes symptomatic when the amyloid has spread beyond the vessels into interstitium.
Bleeding may also be present due to an assortment of coagulation abnormalities, most commonly factor X deficiency, but also due to capillary fragility which results in purpura occurring most notably around the eyes and neck.
Diagnosis
The presence of a monoclonal protein in the serum and/or urine and any of the above presenting features should alert the treating physician to the possibility that a patient has AL amyloidosis. The diagnosis is made by biopsying either an affected organ or a more readily available vascular tissue like fat, bone marrow, or gastrointestinal mucosa. If the tissue stains with Congo Red, and there is green birefringence, the patient can be labeled as having amyloidosis. It is important to note that not all amyloid is AL amyloid and not all AL is systemic. Although AL amyloidosis is the most common type, hereditary, senile, and secondary forms exist and should not be confused with AL amyloidosis because of the different therapies indicated. In addition, a minority of patients may have localized amyloidosis, an entity in which the plasma cells producing the amyloidogenic protein are at the site of the amyloid rather than at the bone marrow. The most common sites for localized amyloidosis are the genitourinary tract, the respiratory tract, and the lymph nodes.
Once a diagnosis of amyloidosis is made, all efforts should be made to type the amyloid itself either by immunohistochemistry or direct sequencing of the amyloid itself. Merely documenting a monoclonal protein or monoclonal plasma cells is not sufficient.8,9 DNA based screening for the most common hereditary forms of amyloid may also be a helpful means of exclusion.
Prognosis
Once a diagnosis of AL amyloidosis is made, it is important to define prognosis. The extent of cardiac involvement is by far the most important prognostic parameter in these patients. As early as the 1970s, it was shown that patients presenting with cardiogenic syncope or CHF had median survivals of 4 to 6 months.10
The number of symptomatic organs was also identified as a prognostic factor for patients, especially those undergoing high-dose chemotherapy with hematopoietic stem cell transplantation (HSCT).11 Problems arose with counting numbers of organs because there was no standardized definition of organ involvement until recently.12 Although implementation of standardized definitions of involved organs is a major improvement, these definitions do not provide information about the degree of organ involvement. The survival of someone with advanced cardiac involvement is a fraction of that of an individual with early involvement. Fortunately, the Pavia group and the Mayo group have identified that cardiac biomarkers are powerful predictors of survival both at presentation 13-15 and during treatment with cytotoxic chemotherapy 16 The advantage of the cardiac biomarkers is that they are more readily available and reproducible than echocardiography. As one uses these assays, one must be cognizant that there are two troponin measurements most commonly in use, the troponin T and the troponin I and two brain natriuretic peptide (BNP) assays, the BNP and the N-terminal BNP. The prognostic usefulness holds true regardless of the assay, but the critical thresholds will vary. For example, a conversion factor between BNP and NT-proBNP is: log BNP = 0.28 + 0.66 * log NT-ProBNP.
Other important prognosticators include baseline serum immunoglobulin free light chain(FLC),17 beta-2 microglobulin18 and plasma cell labeling index19.
Non-transplant therapies for AL amyloidosis
The treatment of the systemic amyloidoses varies greatly according to the type of amyloidosis: therefore accurate diagnosis is of vital importance. At present, the best approach to treating amyloidosis remains in the realm of restricting the amount of protein substrate necessary for amyloid fibril formation. For AL amyloidosis, the current therapeutic approach is based on the observation that organ function can be restored if the synthesis of the amyloidogenic precursor is shut down. The aim of therapy is to rapidly reduce the supply of the amyloidogenic monoclonal light chain by suppressing the underlying plasma cell clone, while using supportive measures to sustain the function of the organs involved.20 A consensus panel from the International Society for Amyloidosis established the criteria for hematologic and organ response.12 It has been demonstrated that achievement of a hematologic response was an important predictor of prolonged survival after HSCT for patients with AL. The degree of response is relevant because patients who achieved a complete response survived longer than those who achieved a partial response, thus indicating that hematologic response may be used as an endpoint in trials assessing AL amyloidosis therapies.21-23 The recent, unpublished data of Merlini et al, indicate that the serial concurrent quantification of the FLC and NT-proBNP in patients with cardiac amyloidosis allows titration of the treatment against the clonal plasma cells according to the organ response, thus optimizing the toxicity-benefit ratio and allowing a prompt change of therapy in the case of an inadequate response.
Important progress has been achieved since the mid nineties when a melphalan and prednisone regimen was considered standard therapy.24 This combination induced slow responses, which were rarely complete, in about 25-30% of patients. The advent of high-dose melphalan followed by rescue with autologous HSCT dramatically changed the perspective for AL amyloidosis care demonstrating that complete response can be achieved in a substantial proportion of eligible patients which translated into organ response and significant survival extension 25. Although HSCT represents a breakthrough in the care of AL amyloidosis, its toxicity has limited its benefit to a minority of patients. Therefore, the search for less toxic and rapidly acting regimens continued. A multicenter trial showed that dexamethasone alone achieves a 53% hematological response rate after a median time of 3.4 months, with 24% complete remissions and a treatment related mortality (TRM) of 7%.26 The addition of oral melphalan to dexamethasone (MDex) induced a hematological response in 67% (CR 33%) of AL patients ineligible for HSCT due to advanced disease, in a median time of 4.5 months, with a low TRM of 4%.27 The extended follow-up of the patients treated with MDex shows that the responses were durable with median progression-free survival of 3.8 years and median overall survival of 5.1 years 28.
Although thalidomide is poorly tolerated in AL patients,29,30 its association with dexamethasone as second-line treatment induces a hematologic response in 48% of patients with 19% CR.31 A risk-adapted oral regimen of cyclophosphamide, thalidomide, and dexamethasone (CTD) in patients with AL amyloidosis produced hematologic response in 74% of patients including complete responses in 21% with a TRM of 4%.32 This regimen has the advantage of preserving hematopoietic stem cells for possible subsequent autologous HSCT, but the actual toxicity of the regimen is not well understood since the regimen was not tested as a formal clinical trial. New drugs have been recently adopted from the therapeutic armamentarium for multiple myeloma for the treatment of AL amyloidosis. The Mayo Clinic and the Boston University groups reported that the thalidomide analog, lenalidomide, particularly when used in combination with dexamethasone, in patients with AL amyloidosis, many of whom were previously treated, induced approximately 50% hematologic response with 22% complete responses. Fatigue and myelosuppression were the most common treatment-related adverse events, while thromboembolic complications were the most serious 33,34. These findings indicate that the combination of lenalidomide and dexamethasone represents a valid treatment option. The proteasome inhibitor, bortezomib, is undergoing an international phase I-II dose-escalating trial in previously treated patients, and preliminary results indicate that this drug can be rapidly effective in a significant proportion of patients with manageable toxicity 35-37.
The goal of chemotherapy is a rapid and effective suppression of the synthesis of the amyloidogenic light chain in order to induce a functional improvement of the organ involved with minimal toxicity. The only randomized trial comparing modern therapies is the French Multicentric Trial on HSCT and MDex.38 The trial showed that HSCT was not superior to MDex in a multicenter setting and was associated with lower survival when patients were treated in centers without great experience (see next section for further discussion).
Optimal therapy for AL amyloidosis remains undetermined due to the lack of large comparative randomized clinical trials to support the use of one agent over another. It is likely that in the near future, early diagnosis and combination therapy with alkylating agents associated with dexamethasone, thalidomide and the new drugs lenalidomide and bortezomib, would lead to extended and better life for all AL patients. Supportive therapy is of paramount importance for gaining time while specific therapy takes effect (reviewed in reference 39). Heart 40 or kidney transplantation 41 followed by autologous HSCT has been successfully applied in selected patients.
High-dose Chemotherapy with Peripheral Blood Stem Cell Transplant for AL Amyloidosis
High dose chemotherapy with HSCT for patients with AL amyloidosis began to come into favor in the mid 1990s.11,21,23,42-47 The very first reports were associated with high treatment-related mortality(TRM).43 However, with risk based melphalan dose reduction, more patients became eligible for the procedure. For many it was considered the treatment of choice for patients with AL amyloidosis because amyloidosis centers performing HSCT and delivering 200 mg/m2 of melphalan as conditioning could boost median survivals approaching 8-years.21 Sicker patients who were treated with attenuated doses of melphalan conditioning (100 to 140 mg/m2) could also be brought through the procedure with a TRM of about 14%, but fared less well with regards to hematologic and organ response rates as well as overall survival.21,22
Questions arose regarding selection bias being responsible for superior outcomes,48 but later, a case controlled study demonstrated improved survival in patients receiving high-dose melphalan with HSCT.49 As mentioned, Palladini et al, however, demonstrated that patients too sick for transplant could not only tolerate MDex, but could achieve remarkably high hematologic and organ response rates.27 Lachmann et al had previously shown that 2 to 6 cycles of low dose intravenous melphalan (25 mg/m2) was also associated with an immunoglobulin free light response rate of approximately 50%3.
These findings prompted the Myélome Autogreffe (MAG) and the Intergroupe Francophone du Myélome (IFM) Intergroup to perform a risk adapted multicenter randomized controlled trial of 100 patients, half receiving melphalan and dexamethasone (MDex) and the other half receiving high-dose melphalan with autologous HSCT. Over a 5 year period from 2000 to 2005, 100 patients were enrolled at 29 centers.38 Among the MDex group, one patient died before starting treatment and another died within 3 days of starting treatment with 96% of patients receiving at least one cycle of therapy. Eighty-six percent of patients received 3 or more cycles of therapy with the median number of cycles of 12. In contrast, among the 50 patients randomized to high-dose melphalan, only 37 (74%) received high-dose melphalan. Reasons included refusal (n=1), death before stem cell mobilization (n=1), insufficient stem cell collection (n=2), death during stem cell mobilization (n=4), sudden death after stem cell collection, but before conditioning (n=5). Of the 37 patients receiving high-dose melphalan, 9 died within 100 days of stem cell infusion resulting in a post-randomization early mortality rate of 38%. Conditioning doses of melphalan used were 140 mg/m2 and 200 mg/m2 in 10 and 27 patients, respectively.
The authors addressed hematologic response in 38 MDex and 27 HSCT patients at 100 days, and found that rates were comparable with just over two-thirds of surviving patients achieving hematologic responses. A higher percentage of responses in the HSCT group were complete responses. On an intention to treat basis, however, overall hematologic responses for MDex and HSCT were 52% and 36%, respectively. Moreover, organ responses were seen in 17 and 13 patients, respectively. With 36 months of follow-up for surviving patients, the median survival rates were 57 and 22 months, respectively. When a landmark analysis was performed at 6 months, the overall survival rates between the 2 arms were identical.
So, what conclusions can be drawn from this important, but small, randomized trial? The first is that it confirms the results from Merlini's group that MDex is a highly active treatment for AL amyloidosis patients, including those with high-risk disease.27 It serves as yet another study that highlights the strong association between hematologic response, organ response, and survival. It also confirms that HSCT is associated with exceptionally high TRM when performed outside of transplant centers with amyloidosis expertise.47,50 A major shortcoming of this study, however, is the lack of modern cardiac biomarkers to assure balance between treatment arms. Given the small sample size and the known heterogeneity of the disease, impeccable matching of the most relevant prognostic markers is essential to guarantee that differences and/or similarities between treatment arms are related to treatment effect rather than patient sampling. The fact that half as many patients died before completing 3 cycles of MDex as compared to those dying prior to receiving high-dose melphalan would suggest that such an imbalance may have existed.
Summary
AL amyloidosis is a rare disease in which immunoglobulin light chains are deposited as aggregated interstitial fibrils that can compromise organ function and lead to death. The risks that patients with amyloidosis face include late diagnosis, misdiagnosis of amyloid type, untimely and ineffective therapy and toxicities of therapy. The goals of treatment are (1) reduction or elimination of the amyloid-forming protein, usually a free immunoglobulin light chain measured by the serum free light chain assay (2) support of the patient pending hematologic response and improvement and (3) stabilization of organ function. Whenever possible, patients should be treated on clinical trials.
Figure 2.
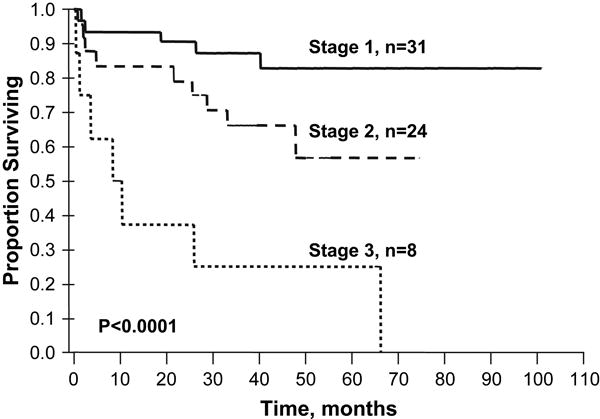
Predicting outcome post autologous peripheral blood stem cell transplant based on cardiac biomarker staging system.
Stage 1, both below biomarkers below cut-off; Stage 2, one of the two biomarkers below cut-off; and Stage 3, both biomarkers above cut-offs. Cut-offs troponin T < 0.035 mcg/L and NT-proBNP < 332 ng/L (39 pmol/L). Results updated from original publication published in Blood. 2004;104:1881-1887. Median follow-up of surviving patients is 35.5 months.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Angela Dispenzieri, Mayo Clinic, Department of Medicine, Division of Hematology, Rochester, MN.
Giampaolo Merlini, Amyloidosis Center, Fondazione IRCCS Policlinico San Matteo, Department of Biochemistry, University of Pavia, Italy.
Raymond L. Comenzo, Memorial Sloan-Kettering Cancer Center, New York, NY
References
- 1.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, G MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Seminars in Hematology. 1995;32:45–59. [PubMed] [Google Scholar]
- 3.Lachmann HJ, Gallimore R, Gillmore JD, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. British Journal of Haematology. 2003;122:78–84. doi: 10.1046/j.1365-2141.2003.04433.x. [DOI] [PubMed] [Google Scholar]
- 4.Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem. 2005;51:878–881. doi: 10.1373/clinchem.2004.046870. [DOI] [PubMed] [Google Scholar]
- 5.Teng J, Turbat-Herrera EA, Herrera GA. Role of translational research advancing the understanding of the pathogenesis of light chain-mediated glomerulopathies. Pathol Int. 2007;57:398–412. doi: 10.1111/j.1440-1827.2007.02116.x. [DOI] [PubMed] [Google Scholar]
- 6.Gertz MA, Kyle RA. Primary systemic amyloidosis--a diagnostic primer. Mayo Clinic Proceedings. 1989;64:1505–1519. doi: 10.1016/s0025-6196(12)65706-1. [DOI] [PubMed] [Google Scholar]
- 7.Palladini G, Kyle RA, Larson DR, Therneau TM, Merlini G, Gertz MA. Multicentre versus single centre approach to rare diseases: the model of systemic light chain amyloidosis. Amyloid. 2005;12:120–126. doi: 10.1080/13506120500107055. [DOI] [PubMed] [Google Scholar]
- 8.Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. doi: 10.1056/NEJMoa013354. [DOI] [PubMed] [Google Scholar]
- 9.Comenzo RL, Zhou P, Fleisher M, Clark B, Teruya-Feldstein J. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood. 2006;107:3489–3491. doi: 10.1182/blood-2005-10-4148. [DOI] [PubMed] [Google Scholar]
- 10.Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271–299. doi: 10.1097/00005792-197507000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Moreau P, Leblond V, Bourquelot P, et al. Prognostic factors for survival and response after high-dose therapy and autologous stem cell transplantation in systemic AL amyloidosis: a report on 21 patients. Br J Haematol. 1998;101:766–769. doi: 10.1046/j.1365-2141.1998.00772.x. [DOI] [PubMed] [Google Scholar]
- 12.Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL). Am J Hematol; a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis; Tours, France. 18-22 April 2004; 2005. pp. 319–328. [DOI] [PubMed] [Google Scholar]
- 13.Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107:2440–2445. doi: 10.1161/01.CIR.0000068314.02595.B2. [DOI] [PubMed] [Google Scholar]
- 14.Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751–3757. doi: 10.1200/JCO.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 15.Dispenzieri A, Gertz MA, Kyle RA, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104:1881–1887. doi: 10.1182/blood-2004-01-0390. [DOI] [PubMed] [Google Scholar]
- 16.Palladini G, Lavatelli F, Russo P, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–3858. doi: 10.1182/blood-2005-11-4385. [DOI] [PubMed] [Google Scholar]
- 17.Dispenzieri A, Lacy MQ, Katzmann JA, et al. Absolute values of immunoglobulin free light chains are prognostic in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2006;107:3378–3383. doi: 10.1182/blood-2005-07-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gertz MA, Kyle RA, Greipp PR, Katzmann JA, O'Fallon WM. Beta 2-microglobulin predicts survival in primary systemic amyloidosis. American Journal of Medicine. 1990;89:609–614. doi: 10.1016/0002-9343(90)90179-h. [DOI] [PubMed] [Google Scholar]
- 19.Gertz MA, Kyle RA, Greipp PR. The plasma cell labeling index: a valuable tool in primary systemic amyloidosis. Blood. 1989;74:1108–1111. [PubMed] [Google Scholar]
- 20.Merlini G, Stone MJ. Dangerous small B-cell clones. Blood. 2006;108:2520–2530. doi: 10.1182/blood-2006-03-001164. [DOI] [PubMed] [Google Scholar]
- 21.Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85–93. doi: 10.7326/0003-4819-140-2-200401200-00008. [DOI] [PubMed] [Google Scholar]
- 22.Gertz MA, Lacy MQ, Dispenzieri A, et al. Risk-adjusted manipulation of melphalan dose before stem cell transplantation in patients with amyloidosis is associated with a lower response rate. Bone Marrow Transplant. 2004;34:1025–1031. doi: 10.1038/sj.bmt.1704691. [DOI] [PubMed] [Google Scholar]
- 23.Perfetti V, Siena S, Palladini G, et al. Long-term results of a risk-adapted approach to melphalan conditioning in autologous peripheral blood stem cell transplantation for primary (AL) amyloidosis. Haematologica. 2006;91:1635–1643. [PubMed] [Google Scholar]
- 24.Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. New England Journal of Medicine. 1997;336:1202–1207. doi: 10.1056/NEJM199704243361702. see comments. [DOI] [PubMed] [Google Scholar]
- 25.Comenzo RL, Vosburgh E, Simms R, et al. Dose-intensive melphalan with blood stem cell support for the treatment of AL amyloidosis: one-year follow-up in five patients. Blood. 1996;88:2801–2806. [PubMed] [Google Scholar]
- 26.Dhodapkar MV, Hussein MA, Rasmussen E, et al. Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood. 2004;104:3520–3526. doi: 10.1182/blood-2004-05-1924. [DOI] [PubMed] [Google Scholar]
- 27.Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936–2938. doi: 10.1182/blood-2003-08-2788. [DOI] [PubMed] [Google Scholar]
- 28.Palladini G, Russo P, Nuvolone M, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110:787–788. doi: 10.1182/blood-2007-02-076034. [DOI] [PubMed] [Google Scholar]
- 29.Dispenzieri A, Lacy MQ, Rajkumar SV, et al. Poor tolerance to high doses of thalidomide in patients with primary systemic amyloidosis. Amyloid. 2003;10:257–261. doi: 10.3109/13506120309041743. [DOI] [PubMed] [Google Scholar]
- 30.Seldin DC, Choufani EB, Dember LM, et al. Tolerability and Efficacy of Thalidomide for the Treatment of Patients with Light Chain-Associated (AL) Amyloidosis. Clinical Lymphoma. 2003;3:241–246. doi: 10.3816/clm.2003.n.005. [DOI] [PubMed] [Google Scholar]
- 31.Palladini G, Perfetti V, Perlini S, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL) Blood. 2005;105:2949–2951. doi: 10.1182/blood-2004-08-3231. [DOI] [PubMed] [Google Scholar]
- 32.Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood. 2007;109:457–464. doi: 10.1182/blood-2006-07-035352. [DOI] [PubMed] [Google Scholar]
- 33.Sanchorawala V, Wright DG, Rosenzweig M, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase II trial. Blood. 2006 doi: 10.1182/blood-2006-07-030544. [DOI] [PubMed] [Google Scholar]
- 34.Dispenzieri A, Lacy MQ, Zeldenrust SR, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109:465–470. doi: 10.1182/blood-2006-07-032987. [DOI] [PubMed] [Google Scholar]
- 35.Wechalekar A, Gillmore J, Lachmann H, Offer M, Hawkins P. Efficacy and Safety of Bortezomib in Systemic AL Amyloidosis - A Preliminary Report. ASH Annual Meeting Abstracts. 2006;108:129. [Google Scholar]
- 36.Kastris E, Anagnosopoulos A, Roussou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. doi: 10.3324/haematol.11325. In press. [DOI] [PubMed] [Google Scholar]
- 37.Reece DL, Sanchorawala V, Hegnebar U, et al. Phase I/II study of bortezomib in patients with systemic AL-amyloidosis. Journal of Clinical Oncology. 2007;25:8050a. [Google Scholar]
- 38.Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357:1083–1093. doi: 10.1056/NEJMoa070484. [DOI] [PubMed] [Google Scholar]
- 39.Merlini G. AL amyloidosis: therapeutic strategies 2004. Hematology (Am Soc Hematol Educ Program) 2004:261–269. [Google Scholar]
- 40.Gillmore JD, Goodman HJ, Lachmann HJ, et al. Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood. 2006;107:1227–1229. doi: 10.1182/blood-2005-08-3253. [DOI] [PubMed] [Google Scholar]
- 41.Leung N, Griffin MD, Dispenzieri A, et al. Living donor kidney and autologous stem cell transplantation for primary systemic amyloidosis (AL) with predominant renal involvement. Am J Transplant. 2005;5:1660–1670. doi: 10.1111/j.1600-6143.2005.00920.x. [DOI] [PubMed] [Google Scholar]
- 42.van Buren M, H RJ, V LF, V FJ, L HM. Clinical remission after syngeneic bone marrow transplantation in a patient with AL amyloidosis. Annals of Internal Medicine. 1995;122:508–510. doi: 10.7326/0003-4819-122-7-199504010-00005. [see comments]. Comment in: Ann Intern Med 1995 Oct 1;123(7):553. [DOI] [PubMed] [Google Scholar]
- 43.Moreau P, Milpied N, de Faucal P, et al. High-dose melphalan and autologous bone marrow transplantation for systemic AL amyloidosis with cardiac involvement. Blood. 1996;87:3063–3064. [PubMed] [Google Scholar]
- 44.Guillaume B, S N, J M, C JP, F A. Allogeneic bone marrow transplantation for AL amyloidosis. Bone Marrow Transplantation. 1997;20:907–908. doi: 10.1038/sj.bmt.1700983. [DOI] [PubMed] [Google Scholar]
- 45.Comenzo RL, Vosburgh E, Falk RH, et al. Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light-chain) amyloidosis: survival and responses in 25 patients. Blood. 1998;91:3662–3670. [PubMed] [Google Scholar]
- 46.Gertz MA, Lacy MQ, Dispenzieri A. Myeloablative chemotherapy with stem cell rescue for the treatment of primary systemic amyloidosis: a status report. Bone Marrow Transplantation. 2000;25:465–470. doi: 10.1038/sj.bmt.1702178. [DOI] [PubMed] [Google Scholar]
- 47.Goodman HJ, Gillmore JD, Lachmann HJ, Wechalekar AD, Bradwell AR, Hawkins PN. Outcome of autologous stem cell transplantation for AL amyloidosis in the UK. Br J Haematol. 2006;134:417–425. doi: 10.1111/j.1365-2141.2006.06204.x. [DOI] [PubMed] [Google Scholar]
- 48.Dispenzieri A, Lacy M, Kyle RA, et al. Eligibility for hematopoietic stem-cell transplantation for primary systemic amyloidosis is a favorable prognostic factor for survival. J Clin Oncol. 2001;19:3350–3356. doi: 10.1200/JCO.2001.19.14.3350. [DOI] [PubMed] [Google Scholar]
- 49.Dispenzieri A, Kyle RA, Lacy MQ, et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral blood stem cell transplantation: a case-control study. Blood. 2004;103:3960–3963. doi: 10.1182/blood-2003-12-4192. [DOI] [PubMed] [Google Scholar]
- 50.Vesole DH, Perez WS, Akasheh M, Boudreau C, Reece DE, Bredeson CN. High-dose therapy and autologous hematopoietic stem cell transplantation for patients with primary systemic amyloidosis: a Center for International Blood and Marrow Transplant Research Study. Mayo Clin Proc. 2006;81:880–888. doi: 10.4065/81.7.880. [DOI] [PubMed] [Google Scholar]