

Abstract
Recent experimental evidence has shown that application of certain neurotrophic factors (NTs) to the developing primary visual cortex prevents the development of ocular dominance (OD) columns. One interpretation of this result is that afferents from the lateral geniculate nucleus compete for postsynaptic trophic factor in an activity-dependent manner. Application of excess trophic factor eliminates this competition, thereby preventing OD column formation. We present a model of OD column development, incorporating Hebbian synaptic modification and activity-driven competition for NT, which accounts for both normal OD column development as well as the prevention of that development when competition is removed. In the “control” situation, when available NT is below a critical amount, OD columns form normally. These columns form without weight normalization procedures and in the presence of positive inter-eye correlations. In the “experimental” case, OD column development is prevented in a local neighborhood in which excess NT has been added. Our model proposes a biologically plausible mechanism for competition between neural populations that is motivated by several pieces of experimental data, thereby accounting for both normal and experimentally perturbed conditions.
A central question in neuroscience concerns the degree to which environmental influences modify neural architecture and function. The ocular dominance (OD) columns of primary visual cortex (V1) is a model system that has been extensively studied due to the marked effects of environment on both anatomy and physiology. These columns consist of cells receiving projections from the lateral geniculate nucleus (LGN), which in turn receive inputs from each eye (1). Around or before birth, LGN afferents subserving each eye consist of overlapping projections, so that all cortical cells are binocular (i.e., receiving inputs from each eye). During development, these afferents segregate, so that most layer IV cells within an OD column receive thalamocortical afferents predominantly from one eye, whereas cells in the adjacent column receive inputs from the opposite eye. This pattern of connectivity is found in many species, such as cats, monkeys, and humans.
Several lines of experimental data (refs. 2–9; for a review, see ref. 9) and theoretical modeling (refs. 10–12), for review, see ref. 13) suggest that OD column development depends on activity-dependent competition among axons from the LGN. Blocking activity in the retina prevents OD column formation (14), and synchronous stimulation of afferent connections increases the percentage of binocularly driven cells (8). Monocular deprivation results in the expansion of open-eye columns at the expense of closed eye columns (4, 6), whereas application of the N-methyl-d-aspartate receptor blocker 2-amino-5-phosphonovaleric acid (15) or blockade of cortical activity with tetrodotoxin (16) prevents this plasticity. These results all suggest a Hebbian mechanism of synaptic plasticity, combined with competition among afferents to maintain connections to cortex. To simulate this competitive aspect, previous models have generally normalized total synaptic strength—i.e., the total amount of the incoming synaptic strengths (or weights) to a cortical cell is constrained to remain constant (17, 18) (although see ref. 19 for an exception). This computation is an abstraction used to enforce competition (as these modelers recognized), but leaves open the issue as to how this competition is actually implemented biologically. Therefore, the question remains: for what entity are the presynaptic afferents competing, and how does the advantage of one afferent over another accrue with activity and current state?
One plausible hypothesis is that afferents compete for a limited supply of post-synaptic trophic factor to maintain connectivity to cortical cells (20-23). Recent experimental evidence has shown that the infusion of excess amounts of certain neurotrophins (NTs), such as brain-derived neurotrophic factor (BDNF) and NT-4/5, prevents OD column development (24). One interpretation of these data is that LGN axons compete for NT, such that in normal animals afferents from one eye win this competition at the expense of the opposite eye. Application of excess trophic factor eliminates this competition, thereby permitting connections from both eyes to remain and preventing the OD column segregation.
We present a model of OD column development incorporating Hebbian synaptic modification and competition among afferents for NT. The model accounts for the development of OD columns and the prevention of that development with application of excess amounts of exogenous NT. The model is based on three essential hypotheses. (i) Synaptic strengths increase due to Hebbian long-term potentiation (LTP), a LTP-like phenomenon, and decrease due to heterosynaptic long-term depression (LTD), a LTD-like phenomenon (the terms LTP and LTD are to be interpreted broadly, not necessarily referring to any specific brain area or experimental protocol). (ii) Positive feedback exists between the rate of LTP and the rate of trophic factor uptake. The more trophic factor accumulated at a synapse, the higher its rate of connection strength increase (LTP); in turn, the higher the synaptic weight, the faster the uptake of trophic factor. (iii) Afferents compete for a limited amount of trophic factor. Computer simulations show that OD columns develop from random initial conditions via spontaneous symmetry-breaking when the amount of postsynaptic trophic factor is below a critical amount; that columns develop with positive inter-eye correlations without weight normalization procedures; and that excess NT applied in a local area of cortex prevents OD column development. The key features of the model (Hebbian weight changes, positive feedback between connection strength increase and NT uptake, and a limiting supply of cortical trophic factor) are motivated by specific pieces of experimental evidence and provide a novel mechanism of competition between neural populations.
METHODS
Model Derivation.
The simulated cells in the network are standard artificial neural network units, with an activation value representing average firing rate, and connection strengths representing peak postsynaptic potentials generated by afferent inputs. Temporal dynamics of cellular activation are not included in the model, consistent with the assumption that the time averaged activity is the important variable in the formation of OD columns. Inputs are modeled as a single continuous variable from each eye. While a more realistic simulation consisting of many inputs from each eye may be important for such phenomena as the development of individual spatial receptive fields or orientation preferences, the model assumes that the gross correlative activity within and between each eye is important for driving OD segregation (8). The model consists of a sheet of cortical neurons receiving inputs from each eye (see Fig. 1a). Cortical neurons interact via center-surround connectivity. The neurons in the model are linear units:
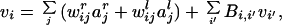 |
1 |
where vi is transmembrane voltage of the ith cortical unit (we use the terms postsynaptic voltage and firing rates interchangeably due to the linearity of the units); wijr,l and ajr,l are, respectively, synaptic strength to the ith cortical unit from the jth thalamic unit from the right or left eye, and activity in the jth thalamic input of the right or left eye; and Bi,i′ and vi′ are, respectively the connection strength from and activity in the i′ cortical unit. In all that follows, j will be dropped because we consider a single thalamic input. Rearranging Eq. 1, we obtain:
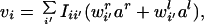 |
2 |
where I = (1 − B)−1 is the intracortical interaction function (1 is the identity matrix). We will derive the equations controlling synaptic strength and trophic factor levels for the afferent from the right eye; the identical equations hold for the left eye with r′ and l′ values interchanged.
Figure 1.

(a) Schematic of model. Cortex is represented by a 30 × 30 grid of units. Each thalamic input is represented by a single cell, thus the correlation matrix in Eq. 11 is a 2 × 2 matrix, given by Cll = Crr = 0.9, Clr = Crl = 0.3. The inset shows the interaction function I between cortical cells, implemented as a difference of gaussians: I(x) = Imaxe−(x/χ1)2 − Imine−(x/χ2)2, where Imax = 1.0, Imin = 0.15, χ1 = 1.3, χ2 = 2.6, and x is the cortical distance between units. (b) Schematic showing single cortical cell receiving inputs from each eye. The connection from each eye has a fixed amount of total synaptic material. Each cortical cell has a fixed amount of trophic factor to distribute over its inputs.
Each cortical cell has a fixed pool of trophic factor to distribute over all thalamic inputs, whereas each individual connection has a fixed amount of material from which to add connection strength (Fig. 1b). Intracortical connections remain fixed for these simulations. The maximum level of thalamocortical connection strength is arbitrarily set to 1 for all synapses:
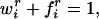 |
3 |
where wir is the current amount of connection strength, and fir is the free store of synaptic raw material still available at a single synaptic locus. We would not make any claims about the validity of such a “free store” per se, but this formulation will have the effect of keeping each synaptic weight between zero and some maximal value, here set to 1 (note that constraining the weights to remain between 0 and some maximal value is in fact necessary for biological plausibility). This will implement a “soft” constraint in that the weights approach their limiting values asymptotically. Trophic factor is dealt with similarly, but here a fixed amount of trophic factor is available postsynaptically for distribution over all incoming synapses. The total amount of trophic factor at the ith cortical unit available to be distributed over inputs is Ni, and equals the sum of the amount currently taken up by the right and left eye afferents, nir + nil, and the free trophic factor left at the ith cortical unit, Nif. Thus,
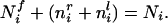 |
4 |
We formulate the equations in terms of mass–action kinetics as a mathematical convenience, not making any claims about the necessity of such a formulation. The free synaptic material is converted to connection strength by the simple kinetic scheme:
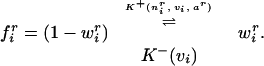 |
5 |
The rate constants are functions depending on the potential, the inputs, and the current amount of synaptic trophic factor. The trophic factor uptake dynamics obey a simple kinetic equation as well:
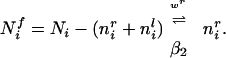 |
6 |
We hypothesize that the stronger the weight, the faster the rate at which NT is taken up into afferents. This feature is motivated by evidence that trophic factor can be released in an activity-dependent manner from application of glutamate (25) (see Discussion). Release of trophic factor back to the free pool is just a constant, β2. The rate “constants”, K±(⋅) and wir, are actually functions of several variables or dynamic variables. The forward rate constant is a simple Hebbian rule, modulated by the current amount of NT:
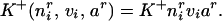 |
7 |
The first term is a constant (to correct for dimensionality, and will be dropped). The last two terms are the product of postsynaptic activity of the ith unit and presynaptic activity (viar), implementing the simplest form of the Hebbian rule (LTP). These are modulated by the current amount of trophic factor taken up at that synapse (nir). Thus the more trophic factor at a synaptic locus, the faster its increase in connection strength. This dependence of LTP on trophic factor is motivated by recent experimental work (26) (see Discussion). Using the definition of vi from Eq. 2, and using a standard averaging procedure over the input ensemble (see refs. 10 and 27 for justification), we obtain:
 |
8 |
for the rate at which synaptic strength from the right eye to the ith unit increases. (Note that because the temporal dynamics of nir are on the same scale as that of wir, the averaging procedure, which in previous papers was used only for changes in the weights, is still valid.) Here, C is the correlation between inputs ar,l, so Crr = 〈arar〉, Cll = 〈alal〉, and Crl = Clr = 〈aral〉. Note that C > 0 always, because ar,l > 0. The backward rate constant depends only on the postsynaptic potential vi. This is an approximation of heterosynaptic LTD (for a review, see ref. 28), as it will tend to dominate the LTP term when presynaptic activity is low and postsynaptic activity is high:
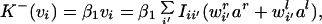 |
9 |
which after averaging is
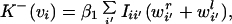 |
10 |
with all constants absorbed into β1. Substituting Eq. 8 and Eq. 10 into the equations describing mass-action kinetics in Eq. 5, we obtain:
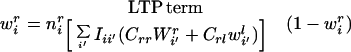 |
11 |
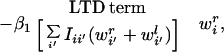 |
the equation describing the change in synaptic strength to the ith cortical unit from the right eye. The identical equation describes the change in synaptic strength from the left eye, with r′ and l′ values interchanged. The first term is the rate of connection strength increase, in which the term representing LTP is positively modulated by the amount of trophic factor currently taken up at that synaptic locus. The second is the rate of connection strength decrease, or LTD. Similarly, the equation describing the kinetics for trophic factor can be derived from Eq. 6:
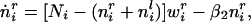 |
12 |
with the symmetric equation for the input from the left eye. The rate of trophic factor uptake is positively modulated by the current synaptic strength and limited by the amount of free postsynaptic trophic factor still available. These coupled equations determine the dynamics of connection strength and trophic factor at each synaptic locus.
Simulation Procedures.
The above differential equations were solved numerically using Euler’s method with a step size between 0.05 and 0.1. To eliminate edge effects, periodic boundary conditions were used at the borders of the cortical sheet, which was 30 × 30 units in size. For all simulations, the simulation was stopped when the percent change of the weights went below some tolerance level. That is, if w(t + 1) = w(t) + δw(t), simulations were stopped when
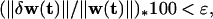 |
13 |
where ∥w(t)∥ = ∑j(t) | wj(t) | (j running over all weights in the net) and ɛ = 0.1. Simulations generally ran for between 1,000 and 3,000 iterations. One nonlinearity was inserted into Eq. 11 order to maintain biological realism and numerical stability. The LTP or LTD terms were clipped at zero so that both numbers were always positive; thus the first term always increases the weights and the second term always decreases them (or they are zero).
A bifurcation diagram for a single cortical cell was constructed numerically using the program xppaut (this software by G.B.E. is available to download at http://www.pitt.edu/~phase). Simulations of the full network were run to determine the model’s performance under two conditions: the “control” condition, in which all cortical cells had a low, equal amount of NT available, and the “experimental” condition, in which an excess amount of NT was added in a limited area. This was modeled by imagining a point source of NT added to the intrinsic NT available in the center of the cortical sheet; this amount then decayed from the point source with distance and was added to each cells’ available NT. OD was measured by simply subtracting the weight from one eye from the weight from the other; thus if ODi = wir − wil, then −1 < ODi < 1, and a cell is completely monocular if ODi = ±1 (weight from one eye is 1, weight from other eye is 0), and completely binocular if ODi = 0 (weight from each eye is equal).
RESULTS
We first present a bifurcation diagram for a single cortical cell receiving inputs from each eye (Fig. 2), which demonstrates the change in solutions as a function of trophic factor (in what follows, when we are discussing a single cortical cell, the i subscripts will be dropped). Plotted on the abscissa is the total amount of trophic factor available; on the ordinate is the weight value from one eye at steady-state. Solid lines are stable solutions at steady-state, whereas dashed lines are unstable. By symmetry, the same diagram applies for each eye. For intermediate amounts of trophic factor (Nm < N < Nb in Fig. 2), two stable asymmetric steady-states are seen, each corresponding to the solution in which one eye dominates in the competition (branches labeled M1 and M2). It can be shown analytically that the monocular solution (e.g., wr = wmon, nr = nmon, wl = 0, nl = 0) is the only stable solution in this range. In other words, whenever wr is on the Ml branch, wl is on the M2 branch, and vice versa. For very low values of NT, (N < Nm) a branch goes below zero; however, for this parameter range, it can be shown that only the zero solution (i.e., wr = wl = nr = nl = 0) is stable. As NT is increased, but below the upper critical amount (Nm < N < Nb), the asymmetric steady-state increases, corresponding to increasing synaptic strength of the winning eye as trophic factor increases. Finally, a critical value is reached (N = Nb), and a bifurcation occurs. At this bifurcation, the symmetric steady-state becomes stable (the branch labeled B). This corresponds to a binocular cell in which weights from both eyes are equal. Thus for levels of NT above the critical amount (N > Nb), the binocular solution will become stable. The two asymmetric solutions also seen for N > Nb (one of which is below zero) are separated from the symmetric steady-state by unstable branches. Due to these unstable branches, for realistic initial conditions these branches are never reached, and trajectories must converge to the symmetric steady-state. The quantitative difference between the critical point for a single cell shown in the bifurcation diagram and the critical point for the cells in the full network in the following figures is due to the intracortical interaction function. We posit that the results in ref. 24 reflect the fact that the developing neocortex has been flooded with trophic factor, putting it above the critical amount and making the binocular solution stable in that area.
Figure 2.

Bifurcation diagram showing different steady-states of weight from one eye as a function of amount of postsynaptic trophic factor available (bifurcation diagram for the other eye is the same by symmetry). Stable steady-states are given by solid lines, and unstable steady-states given by dashed lines. See text for explanation. Parameters are same as in Figs. 3 and 4, (Crr = Cll = 0.9, Clr = Crl = 0.3, β1 = 1.2, β2 = 0.2) except diagram is for a single cortical cell (i.e., i = 1).
Next, we present results from a “control” experiment of a full cortical network. Simulations demonstrate spontaneous symmetry-breaking from random initial conditions to form OD columns. A representative experiment is shown in Fig. 3a. Cortical cells begin with small, equal initial weights from each eye, perturbed by a small amount of noise. Likewise, individual connections begin with a small, approximately equal amount of trophic factor. Each cortical cell has the same amount of trophic factor to distribute over incoming afferents, which is set below a critical amount. The results are robust with respect to random initial conditions. We have also examined the effects of changing inter- and intra-eye correlations, the rate constants, and the default NT amount, generally with similar results. For all correlations values used, there exist some parameter ranges of the rate constants and default NT amounts for which a normal qualitative pattern forms, and this range is often broad. As inter-eye correlations approach intra-eye correlations, the ranges narrow, as would be expected. In the ranges in which OD columns form, changing parameters alters the magnitudes of the final weights. Varying the intracortical interaction function parameters can change the results qualitatively for certain parameter values. Specifically, if there is too much inhibition in the network (i.e., if the magnitude or width of the inhibitory gaussian is too large), then all weights decay to zero, or there is no change from the initial conditions because the LTP and LTD terms are clipped at zero (see Methods). If there is too much excitation in the network, then most cells come to be dominated by one eye, with small pockets dominated by the other eye. For a reasonable range of values, however, individual cells are driven to become monocular, whereas the intracortical connectivity acts to bias nearby cells to have similar eye preference, and distant cells to have opposite eye preference, patterning the cortex into OD columns. Most cells in the cortical sheet are completely monocular in that the nondominant input has a strength of zero; grey scale thus demonstrates the strength of the input from the dominant eye. This is further demonstrated by Fig. 3b, which plots the weights from the right and left eyes to each cortical unit in a slice through the center row of the sheet (the 15th row).
Figure 3.

(a) Simulation results after convergence to steady-state with small, randomly perturbed initial conditions. That is wir = wil = nir = nil = 0.1 + ρ at t = 0, where ρ is a uniformly distributed random number between −0.01 and 0.01. The total amount of trophic factor available at each cortical cell was set to 3.0 (Ni = 3.0 in Eq. 12). A gray scale illustrates synaptic strength from each eye for each cell, from black (all synaptic strength from one eye) to gray (equal synaptic strength from each eye) to white (all synaptic strength from other eye). The scale bar runs from −1 to 1. All final synaptic strengths are between 0 and 1. Most cells are monocular in that synaptic strength from the losing eye is zero; grey scale thus measures connection strength from winning eye. (b) Synaptic strength from each eye to each cortical unit in a slice through the center of the sheet (row 15). Data from simulation depicted in a.
To model the application of exogenous trophic factor (the experimental condition), the amount of NT available in a particular cortical area is increased (see Methods for specifics of how this was done). Parameters such as the height of the gaussian representing concentration of NT at the cannula were varied, as well as the diffusion constant controlling the spatial decay of the exogenous NT, and the interactions between these values with the above-mentioned parameters (input correlations, rate constants, default NT values) were examined. In general, the width and magnitude of the excess NT diffusion could affect the outcome. For a range of parameters, the outcome was similar to that shown in Fig. 4a, with the weights plotted for each cortical unit in Fig. 4b. This experiment has the same parameter values as Fig. 3, but with an excess amount of NT added. Notice the area in the middle where the OD columns have been prevented. The value of the weights in this area is similar to that in the monocular areas, but now both weights have the same high value. Thus binocular cells now form throughout the region of excess NT, providing a theoretical mechanism to explain the results in ref. 24. It explains further why OD columns remain outside the local region in which excess NT is present. For some parameter ranges we have tried, excess NT can actually increase monocularity (i.e., strength of the winning synapse) either completely throughout the area in which NT has been increased, or around the periphery where NT is in an intermediate range. This is suggested by the bifurcation diagram of Fig. 2, in which synaptic strength from the winning eye increases with increasing NT below the critical value. This would suggest that increasing levels of exogenous NT in the cortex could have complex effects depending on the strength and spatial extent of intracortical weights, the input correlations, and the magnitude and spatial decay of excess NT around the cannula. However, the fact that there is good qualitative agreement (such as Fig. 4) between the model and the results in ref. 24 confirms its sufficiency to account for the control and perturbed experimental conditions.
Figure 4.

(a) Same parameters as before, but amount of available trophic factor was the default value of 3.0 plus the exogenous trophic factor added at the cannula in the center which diffused to the cortical locus. Thus, Ni = 3.0 + Nsourcee−(x/χ3)2, where Nsource = 20.0, χ3 = 4.0, and x is the cortical distance between the ith unit and the center of the sheet (the 15th × 15th cortical unit). The gray scale is the same as in Fig. 3. OD columns are eliminated in local patch where excess trophic factor was available. (b) Synaptic strength from each eye to each cortical unit in a slice through the center of the sheet (row 15). Data from simulation depicted in a.
DISCUSSION
The present model suggests a mechanism by which interactions between activity-dependent weight modification and competition for trophic factor generate monocular cells. The essential features are as follows: (i) a positive feedback interaction between the rates of connection strength increase and neurotrophin uptake, and (ii) stabilization of this feedback by competition for a fixed amount of trophic factor. When available postsynaptic trophic factor is below a critical amount, competition drives one eye’s afferents to eliminate the competing eye’s inputs. Addition of excess trophic factor eliminates the competition, thereby preventing monocular cell development.
Many previous models have accounted for OD column development with negative or zero between-eye correlations. Efforts that have modeled the more plausible situation of small positive correlations between the two eyes have relied on weight normalization schemes to enforce competition (see ref. 17 for some of the theoretical issues involved). This computation is an abstraction that leaves open the question of a mechanism for competition. The present model proposes such a mechanism and is motivated by certain biological evidence. By this mechanism, OD columns form with positive inter-eye correlations and without such weight normalization schemes. The role of Hebbian learning in OD column formation has substantial evidence (for a review see ref. 9), and has been central to many previous models, as noted above. Our hypothesized mechanism of competition, namely the positive feedback relationship between trophic factor uptake and synaptic strength increase, is motivated by specific pieces of experimental evidence and accounts for normal and experimentally perturbed conditions.
The positive influence of trophic factor on LTP has been suggested by experiments in the rat hippocampal system in knockout mice (26). These researchers found that mice with the gene for BDNF specifically knocked out showed a severe decrease in the magnitude of and ability to induce LTP in the hippocampus. Subsequent work has shown a restoration to normal of knockout mice with exogenously replaced BDNF (29). Because the heterozygote mice were close to the homozygotes, this suggests that a threshold amount of BDNF is necessary for normal LTP. In our model, trophic factor affects LTP in a linear fashion. However, this linearity is not necessary, and in fact is a weaker assumption than the more biologically realistic nonlinear relation. We have run preliminary simulations for a single cell in which the amount of NT at a synapse is first passed through a sigmoid function before multiplying the LTP term, thus approximating the threshold relationship. These have yielded good results. Therefore, we would not make the claim that trophic factor must linearly affect LTP induction and/or magnitude. The essential feature is that there is a monotonic influence of NT on LTP. This hypothesized mechanism leads to falsifiable predictions. For example, if all thalamocortical synapses in normal animals were to have sufficient NT levels for robust LTP, this would invalidate the model.
Furthermore, this line of reasoning leads to the following prediction. During the OD segregation process, when inputs from both eyes still remain, the eye that is winning the competition would have taken up more NT than the losing eye. Because NT positively affects LTP induction and/or magnitude, LTP should be more easily induced and/or be of higher magnitude in the afferents from the winning eye compared to the afferents from the losing eye. Some possibly contradictory evidence suggests that in the hippocampus, weaker synapses are more easily potentiated via LTP, while stronger synapses are more easily depotentiated via LTD (for a review see ref. 30). However, in that system there is no reason to think that two or more populations compete for a limiting supply of trophic factor, so that all synapses may have more or less equal levels of NT. In our model, for a fixed level of NT at a synapse, weaker synapses are indeed potentiated more easily than strong ones, and stronger synapses depotentiated more easily than weak ones (due to the soft constraint terms). The model suggests that, if NT can affect LTP as hypothesized, the differences in NT levels should induce differences in LTP induction and/or magnitude between competing populations.
In addition, for the other half of the positive feedback loop, evidence has shown increased release of trophic factor from postsynaptic cells with depolarization (25). (While this study examined effects of depolarization on release of nerve growth factor, we assume for the sake of argument that this is representative of BDNF.) Motivated by this result, we hypothesize that NT release might further be confined to individual synapses in an activity-dependent manner. For example, if NT were differentially released from potentiated synapses due to a higher local depolarization, then inputs from the winning eye could take up NT at the expense of the losing eye. If NT diffused too quickly and/or too far from potentiated synapses subserving the winning eye to weaker synapses subserving the losing eye, this would invalidate this mechanism. Alternatively, if presynaptic TrkB receptors (BDNF- and NT-4/5-specific receptors) were regulated, either in number or affinity, in proportion to the synaptic strength at a particular focus, this could also implement the NT uptake dependence on synaptic weight. To our knowledge, no evidence exists either for or against this second possibility.
Finally, the assumption that there is a limiting supply of NT in the normal developing neocortex, and that this level is similar across layer IV cells, is reasonable. The existence of relatively small amounts of TrkB receptors on thalamocortical afferents is consistent with this assumption. Evidence has suggested the BDNF levels can be altered by afferent neurotransmitter (31) and environmental conditions (32). Our model would predict that any up-regulation that occurs under physiological conditions is still beneath the critical value necessary for thalamocortical segregation. Another possibility is that regulation of BDNF levels occurs on a much slower time scale than changes in weights, and could be accompanied by or induce changes in parameters controlling NT uptake. While this would require substantial revision of the model, it could be handled conceptually within this framework.
A significant number of theoretical models have addressed OD column formation (for a review see ref. 15). The current model has several features in common with these previous efforts, namely Hebbian learning rules and center-surround intracortical interactions. An analysis comparing three related models (10, 12, 33) in terms of the bifurcations which each can admit for a single cortical cell has been done (34). In each of these models, there are terms identical or close to our LTP and LTD terms. The model in ref. 10 has no stable fixed points, and hence cannot admit a true bifurcation. That model, however, addressed early development when linear dynamics dominate, and can admit a “bifurcation-like” phenomenon in that the binocular eigenvector can grow slower or faster than the monocular eigenvector depending on a single parameter related to our N. However, if soft constraints are added [multiplying the LTP term by (1 − wr) and the LTD term by wr], the binocular solution is the only stable steady-state, and a monocular cell cannot form. Conversely, in the model in ref. 33, a monocular cell is the only stable steady-state, and a binocular cell cannot form. These results suggest that the latter two models are insufficient to account for the change from a monocular to a binocular steady-state with changes in a single parameter (presumed to correspond to NT levels). Finally, the model in ref. 12 does admit a bifurcation in which both the monocular and binocular solutions exist and their stability can change as a function of a single parameter. For the two input case, this model turns out to be formally identical to Miller’s, but with soft constraints implemented differently [by multiplying the entire LTP and LTD difference by wr(1 − wr)]. Therefore, these two models could qualitatively describe the change of stability from a monocular to a binocular solution with increases in a single parameter. It might be interesting to attempt to re-interpret these models in order to suggest an alternative mechanism by which segregation is prevented by excess NT.
The model is motivated by specific neurobiological data, and its hypotheses are testable and generate results consistent with biological observations. In particular, OD columns develop with a limited supply of postsynaptic trophic factor, and do not develop in the presence of excess NT. Furthermore, OD columns develop with positive inter-eye correlations through plausible biological mechanisms, including Hebbian learning rules, competition for a limited supply of trophic factor, enhancement of connection strength increase by trophic factor uptake, and enhancement of trophic factor uptake with increased connection strength. The most important prediction from our model concerns the positive feedback relation between LTP and NT. These are the essential features of our model by which the competitive process is implemented, and provide a novel mechanism for competitive interactions in the central nervous system.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: OD, ocular dominance; LGN, lateral geniculate nucleus; NT, neurotrophin; BDNF, brain-derived neurotrophic factor; LTP, long-term potentiation; LTD, long-term depression.
References
- 1.Rakic P. Nature (London) 1976;261:467–471. doi: 10.1038/261467a0. [DOI] [PubMed] [Google Scholar]
- 2.Shatz C J, Stryker M. J Physiol (London) 1978;281:267–283. doi: 10.1113/jphysiol.1978.sp012421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shatz C J, Lindstrom S, Wiesel T. Brain Res. 1977;131:103–116. doi: 10.1016/0006-8993(77)90031-2. [DOI] [PubMed] [Google Scholar]
- 4.Wiesel T N, Hubel D. J Neurophysiol. 1965;28:1029–1040. doi: 10.1152/jn.1965.28.6.1029. [DOI] [PubMed] [Google Scholar]
- 5.Hubel D, Wiesel T, LeVay S. Philos Trans R Soc London B. 1977;278:377–409. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]
- 6.LeVay S W, Torsten N, Hubel D H. J Comp Neurol. 1980;191:1–51. doi: 10.1002/cne.901910102. [DOI] [PubMed] [Google Scholar]
- 7.LeVay S, Stryker M, Shatz C J. J Comp Neurol. 1978;179:223–244. doi: 10.1002/cne.901790113. [DOI] [PubMed] [Google Scholar]
- 8.Stryker M P, Strickland S. Invest Opthal Vis Sci (Suppl) 1984;25:278. (abstr.). [Google Scholar]
- 9.Shatz C. Neuron. 1990;5:745–756. doi: 10.1016/0896-6273(90)90333-b. [DOI] [PubMed] [Google Scholar]
- 10.Miller K D, Keller J, Stryker M. Science. 1989;245:605–615. doi: 10.1126/science.2762813. [DOI] [PubMed] [Google Scholar]
- 11.Obermeyer K, Blasdel G, Schulten K. Phys Rev A. 1992;45:7568–7589. doi: 10.1103/physreva.45.7568. [DOI] [PubMed] [Google Scholar]
- 12.Swindale N. Proc R Soc London B. 1980;208:243–264. doi: 10.1098/rspb.1980.0051. [DOI] [PubMed] [Google Scholar]
- 13.Swindale N. Network. 1996;7:161–247. doi: 10.1088/0954-898X/7/2/002. [DOI] [PubMed] [Google Scholar]
- 14.Stryker M P, Harris W A. J Neurosci. 1986;6:2117–2133. doi: 10.1523/JNEUROSCI.06-08-02117.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleinschmidt A, Bear M F, Singer W. Science. 1987;238:355–358. doi: 10.1126/science.2443978. [DOI] [PubMed] [Google Scholar]
- 16.Reiter H, Waitzman D, Stryker M. Exp Brain Res. 1986;65:182–188. doi: 10.1007/BF00243841. [DOI] [PubMed] [Google Scholar]
- 17.Miller K D, MacKay D. Neural Comp. 1994;6:100–126. [Google Scholar]
- 18.von der Malsburg C. Kybernetik. 1973;14:85–100. doi: 10.1007/BF00288907. [DOI] [PubMed] [Google Scholar]
- 19.Bienenstock E L, Cooper L N, Munro P W. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berardi N, Cellerino A, Domenici L, Fagiolini M, Pizzorusso T, Cattaneo A. Proc Natl Acad Sci USA. 1994;91:684–688. doi: 10.1073/pnas.91.2.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmignoto G, Canella R, Candeo P, Comelli M C, Maffei L. J Physiol (London) 1993;464:343–360. doi: 10.1113/jphysiol.1993.sp019638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Domenici L C, Cellerino A, Berardi N, Cattaneo A, Maffei L. NeuroReport. 1994;5:2041–2044. doi: 10.1097/00001756-199410270-00013. [DOI] [PubMed] [Google Scholar]
- 24.Cabelli R J, Hohn A, Shatz C. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 25.Blochl A, Thoenen H. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 26.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linsker R. Proc Natl Acad Sci USA. 1986;83:7508–7512. doi: 10.1073/pnas.83.19.7508. , 8390–8394, 8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bear M F, Malenka R C. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 29.Thoenen H. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 30.Abraham W C, Bear M F. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 31.Lindholm D, Castrén E, Berzaghi M, Blöchl A, Thoenen H. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- 32.Castrén E, Zafra F, Thoenen H, Lindholm D. Proc Natl Acad Sci USA. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka S. Biol Cybern. 1991;64:263–272. doi: 10.1007/BF00199589. [DOI] [PubMed] [Google Scholar]
- 34.Harris A E. Ph.D. thesis. Pittsburgh, PA: Univ. of Pittsburgh; 1997. [Google Scholar]