

Abstract
The structure of lactose permease from Escherichia coli in its lipid environment was studied by attenuated total reflection Fourier transform infrared spectroscopy. The protein exhibits an α-helical content of about 65% and about 25% β-sheet. Unusually fast hydrogen/deuterium (H/D) exchange to 90–95% completion suggests a structure that is highly accessible to the aqueous phase. An average tilt angle of 33° for the helices was found with respect to the bilayer normal at a lipid-to-protein ratio of ≈800:1 (mol/mol), and the permease exhibits optimal activity under these conditions. However, upon decreasing the lipid-to-protein ratio, activity decreases continuously in a manner that correlates with the decrease in the lipid order parameter and the increase in the average helical tilt angle. Taken together, the data indicate that the structure and function of the permease are strongly dependent on the order and integrity of the lipid bilayer.
Understanding of structure–function relationships in membrane proteins is limited for two major reasons: (i) most membrane proteins are difficult to crystallize and therefore their x-ray structures have not been determined, and (ii) the physicochemical influence of the surrounding lipid bilayer on the protein is poorly understood on a molecular level. The lactose permease of Escherichia coli is a paradigm for secondary membrane transport proteins and a suitable candidate to approach these problems. The permease has been overexpressed, purified, reconstituted, and shown to catalyze β-galactoside: H+ symport as a monomer (for recent reviews, see refs. 1 and 2). All available evidence indicates that the protein spans the bilayer with 12 transmembrane α-helices. In addition, freeze–fracture electron microscopy reveals a cleft in the protein that might be related to substrate transport, thereby making the protein highly accessible to the surrounding aqueous phase (3).
Employing site-directed and Cys-scanning mutagenesis, as few as four residues in the permease [Glu-269 (helix VIII), Arg-302 (helix IX), His-322 (helix X), and Glu-325 (helix X)] have been shown to be irreplaceable for H+-coupled sugar translocation. Most recently, a molecular mechanism for coupling between H+ and lactose translocation has been presented which assigns roles to the four essential residues and their functional relationship to the substrate translocation pathway (4). A central aspect of the proposed mechanism is substrate-induced movement and protonation of Glu-325 from an ionic interaction with Arg-302 into the lipid environment. In the low dielectric environment, the pKa of Glu-325 increases dramatically, and when substrate is released, the ionic interaction between Glu-325 and Arg-302 is reestablished, resulting in deprotonation of Glu-325. Numerous additional effects of the lipid environment on the functional integrity of the permease have been observed. Phosphatidylethanolamine (PE; 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) has been shown to act as a chaperone for the in vivo assembly of the protein and folding of the epitope for a monoclonal antibody (5, 6). Moreover, the assembled protein requires PE for active transport (7). Taken together, these data point to an important role of the phospholipid environment on the structure and function of lac permease.
In this report, the secondary structure of lactose permease is determined by attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy, and activity is correlated with average α-helical tilt angle and lipid order parameter as a function of the lipid-to-protein ratio. The results suggest a critical lateral lipid packing pressure as an essential prerequisite for proper membrane insertion and function of the protein.
MATERIALS AND METHODS
Permease Preparation.
E. coli XL1blue (Stratagene) containing the plasmid PTC189 + 6His417, which encodes lac permease with 6 His residues at the C terminus, were grown, and membranes were prepared using a French pressure cell as described (8). The membrane fraction was resuspended in 50 mM phosphate buffer (KPi), 200 mM KCl, 0.5 mM Pefabloc (Boehringer Mannheim), 10 mM 2-mercaptoethanol, and 0.25 mg/ml PE/1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (PG; phosphatidylglycerol) (3:1, wt/wt) at pH 7.2 (buffer I) at 1 ml/g original cells (wt/wt). Solubilization was performed by adding n-dodecyl β-d-maltopyranoside (DM) to a final concentration of 2%. The DM extract from 10 g cells was bound to 3.5 ml Ni-NTA superflow (Qiagen, Chatsworth, CA) in batch by shaking for 2 h at 4°C. The nickel resin was washed three to four times with buffer I and once with 50 mM Hepes, 0.008% DM, 10% glycerol, 15 mM imidazole, and 0.1 mg/ml PE/PG (3:1, wt/wt) (pH 7.2). Bound protein was eluted with 50 mM Hepes, 0.008% DM, 10% glycerol, 200 mM imidazole, and 0.1 mg/ml PE/PG (3:1, wt/wt) (pH 7.2). The eluate was dialyzed overnight into 50 mM Kpi, 0.008% DM, and 0.02 mg/ml PE/PG (3:1, wt/wt) (pH 7.5).
Incorporation of Permease into the Lipid Bilayer.
The solubilized, purified permease was added to PE/PG (3:1, wt/wt) and 1% DM (wt/wt) to yield a protein-to-lipid ratio of 1:12 unless otherwise specified. The sample was then incubated on ice for 15 min and rapidly diluted 180-fold into 50 mM KPi (pH 7.5). The sample was stirred for 15 min at room temperature and centrifuged at 158,000 × gmax for 4 h. The pellet was washed once in 50 mM KPi (pH 7.5) and resuspended in a small volume of the same buffer.
IR Spectroscopy.
IR spectra were collected in a Bomem (Quebec) DA3 FTIR spectrometer purged with N2. Typically, interferograms were recorded with 4 cm−1 spectral resolution and processed using Blackman Harris apodization, 1,000 scans were averaged for one spectrum. A modified continuous flow ATR setup (Janos Technology, Townshend, VT) equipped with a wire grid polarizer (Harrick Scientific, Ossining, NY) was used. The sample (250 μl; ≈20 mM permease) was dried on a Germanium (Ge) internal reflection element (25 reflections), and measurements were performed at 20°C using an incident angle of 45°. Perdeuterated buffers were made by lyophilizing the respective buffer, resuspending it in D2O, and adjusting the pD with NaOD or DCl to the desired value (pD = pH + 0.4 pH units).
Data Analysis.
After data acquisition, an automatic baseline correction of the spectra was performed when needed. For orientational studies, the spectra were zeroed at 1,800 cm−1 for the amide I region and at 3,000 cm−1 for the CH stretching region. For second derivative spectra, the Savitzky–Golay algorithm using seven convolution points was employed. To determine order parameters, the electrical field amplitudes on the Ge lipid interface were calculated assuming a thick multilayer lipid film that exceeds the penetration depth of the evanescent field as:
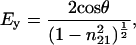 |
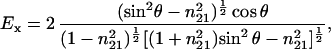 |
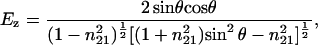 |
where θ is the angle of the incident light (45°) and n21 is the ratio of the refractive indices nlipid = 1.43 and ncrystal = 4.0 (9). The ratio of the integral absorbances A∥/A⊥ was calculated to determine the dichroic ratio (RATR), and the order parameter S was calculated with:
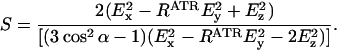 |
For α, the angle between the helix director and the transition dipole, 39° was used (10). Using the calculated order parameter the average helical tilt angle β was determined as:
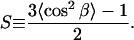 |
Activity Assay.
A counterflow assay (8) was employed to measure transport activity. Briefly, the dried proteolipid film (≈300 μg permease) on the Ge crystal was carefully resuspended in 200 μl of 50 mM KPi, 20 mM lactose, and 1 mM DTT (pH 7.5) and incubated 30 min on ice. After 5- to 10-sec sonification, the suspension was diluted 1:10 into 50 mM KPi, 2 mM MgSO4, and 0.6 mM [1-14C]lactose (pH 7.5) at room temperature. At given time points, 100 μl was filtered through the center of a nitrocellulose filter (pore size, 0.45 μm; Sartorius) and washed immediately with 5 ml of 100 mM KPi, 100 mM LiCl, and 10 mM MgSO4 (pH 5.5). Control points were obtained by diluting the loaded proteoliposomes into the same reaction mixture containing 10 mM β-d-galactopyranosyl-1-thio β-d-galactopyranoside (TDG).
RESULTS
Permease Activity.
The data in Fig. 1 show counterflow activity of lac permease after drying onto the Ge internal reflection element in comparison with a control sample. Surprisingly, no decrease in activity is observed for the dried sample. The time course of the reactions is similar to previously published data (8). It is also noteworthy that a significant amount of bound water is seen in the OH stretching region of the IR spectra (data not shown), indicating that the protein was not completely dehydrated after drying on the Ge element.
Figure 1.

Counterflow activity of rehydrated lac permease. Time course of lactose counterflow in reconstituted proteoliposomes using a protein-to-lipid ratio of 1:12 (wt/wt) corresponding to a stoichiometric ratio of 1:756 (for details, see Materials and Methods). ▪, Proteoliposomes were kept in suspension throughout the experiment; ◊, undehydrated proteoliposomes were assayed in the presence of 10 mM TDG; ○, proteoliposomes were dried onto a Ge internal reflection element and resuspended for counterflow; ▴, proteoliposomes were dried onto a Ge internal reflection element, resuspended for counterflow, and assayed in the presence of 10 mM TDG.
Secondary Structure.
The ATR-FTIR absorbance spectrum of lac permease is dominated by the bands of the lipid ester C=O stretching modes at 1,738 cm−1, amide I and II bands at 1,653 and 1,545 cm−1, and the absorption of the methylene scissoring modes at 1,465 cm−1 and 1,456 cm−1 (Fig. 2). The second derivative of the spectrum reveals more details. In the amide I region between 1,600 and 1,700 cm−1 the band at 1,655 cm−1 clearly indicates α-helical structure as the main component of the protein, which is consistent with the amide II absorption maximum at 1,546 cm−1. In addition, the band at 1,633 cm−1 is indicative of the presence of a significant amount of β-sheet and the bands at 1,695 cm−1 and 1,683 cm−1 show the presence of β- or γ-turns (for band assignments, see refs. 11–14). The fine structure of several small bands that are not assigned arises from side chain contributions of the protein.
Figure 2.

ATR-FTIR spectra of proteoliposomes reconstituted with lac permease in H2O. (A) ATR-FTIR absorbance spectrum obtained with a film of proteoliposomes dried from H2O using parallel polarized light. The dominant bands are the lipid ester C=O stretching vibration at 1,738 cm−1, the amide I and II vibrational modes at 1,653 cm−1 and 1,545 cm−1 and the lipid-methylene scissoring modes at 1,465 cm−1 and 1,456 cm−1. Solid line, functional proteoliposomes with a protein-to-lipid ratio of 1:12 (wt/wt); dotted line, proteoliposomes at a protein-to-lipid ratio of 1:1 (wt/wt) with the spectrum scaled to match the amide II absorption of the solid line. (B) Second derivative spectrum of the solid spectrum in A.
H/D Exchange.
With respect to water accessibility of the peptide backbone 85–90% of the H/D exchange takes place within 1–5 min and about 95% completion is observed after 24 h (Fig. 3). Similar observations were made with the human erythrocyte glucose transporter (15). The major difference between the spectra taken in H2O and D2O is given in the ratios of the amide I to amide II bands. In D2O, the amplitude of the amide I band is smaller compared with the spectrum taken in H2O due to the lack of the overlaying H2O absorption which shifts to 1,215 cm−1 in D2O. Most of the amide II intensity shifts to 1,450 cm−1 in D2O and overlays the methylene scissoring absorption of the lipids that is already present in H2O. However, a small amount of the amide II absorption does not shift, reflecting peptide groups that are inaccessible to D2O. The results clearly demonstrate that most of the protein backbone is highly accessible to bulk water.
Figure 3.

ATR-FTIR spectra of proteoliposomes reconstituted with lac permease in D2O. (A) ATR-FTIR absorbance spectrum of the sample in Fig. 2 rehydrated for 24 h in liquid D2O using parallel polarized light. The dominant bands correspond to the spectrum in Fig. 2A. Most of the amide II intensity is shifted from 1,539 cm−1 to 1,450 cm−1. Note the overall decrease in intensity caused by less contact of the film to the Ge crystal due to swelling of the film in the liquid phase. (B) Second derivative spectrum of the spectrum in A.
Quantification of Secondary Structure.
Deconvolution and band fitting of the amide I absorption in H2O and D2O was performed, and the resulting components were weighed against the total amide I intensity (data not shown). Even though the results of the fitting procedure vary depending on the input parameters, the bands at 1,655 cm−1 and 1,633 cm−1 are clearly identified in all iterations. Calculation of the respective integral intensities for the derived bands suggest an α-helical content of about 65% and a β-sheet content of about 25%. The observed helical content is consistent with previous work obtained from circular dichroism (16) and Raman spectroscopy (17).
Effects of Varying Lipid-to-Protein Ratios.
lac permease was reconstituted into proteoliposomes at increasing lipid-to-protein ratios: 1:1, 3:1, 6:1, 12:1, and 24:1 (wt/wt), or 63:1, 189:1, 378:1, 756:1, and 1512:1 (mol/mol). Dilution of the protein with phospholipid leads to a relative increase of the lipid C=O stretching mode as compared with the amide I and II vibrational modes (see Fig. 2, dotted line). The lipid-to-protein ratios were verified by the method of Tamm and Tatulian (18), and found to be in good agreement with the ratios of the initially prepared samples. Difference dichroism spectra (A∥ − A⊥, data not shown) of the samples reveal a positive dichroism for the amide I band and a negative dichroism for the lipid ester stretching vibration at 1,738 cm−1, indicating a transmembrane orientation of the α helical structure in a bilayer parallel to the Ge internal reflection element. To determine the overall orientation of the protein in the lipid bilayer more accurately, the dichroic ratios of the amide I and the lipid vibrational modes were measured and the average α helical tilt angles were calculated (see Materials and Methods). The dichroism spectra in H2O and in D2O are shown in Fig. 4 A and B, respectively, for a lipid-to-protein ratio of 12:1 (wt/wt), which yield an average α-helical tilt angle of 33°. The difference in the tilt angles obtained in H2O and D2O is due to the superimposed water absorption in the amide I vibrational mode (Fig. 4A). Additional orientational information was obtained by determining the order parameters of the lipids monitored at the methylene symmetric stretching vibration of the fatty acid acyl chains at 2,852 cm−1 (Fig. 4C). At a lipid-to-protein ratio of 756:1 (mol/mol), the membrane lipids assume an order parameter of 0.557. Due to the presence of unsaturated bonds and gauche conformations in the lipid acyl chains, only the order parameter and not the tilt angle of the lipids was determined.
Figure 4.

Polarized ATR-FTIR absorbance spectra. Polarized ATR-FTIR absorbance spectra were obtained with proteoliposomes at a lipid-to-protein ratio of 1:12 (wt/wt) as described. Upper trace, parallel polarized light; bottom trace, perpendicular polarized light. (A) Lipid C=O stretching, amide I, amide II, and methylene scissoring vibrational modes of a spectrum obtained from a film of proteoliposomes dried from H2O buffer. RATR (amide I) = 2.15, order parameter S = 0.102, average α helical tilt angle β = 50°. (B) Spectra obtained on the same film of proteoliposomes immediately after resuspension in D2O buffer in the same spectral region as in A. RATR (amide I) = 2.96, S = 0.537, β = 33°. (C) Methylene stretching modes of the same sample as in B. RATR (sνCH2) = 1.26, S = 0.542.
Correlation between the structural information in different lipid-to-protein ratios to counterflow activity is given in Fig. 5. A dramatic decrease in activity is observed when the lipid-to-protein ratio decreases below 800 lipid molecules per permease molecule. Even more striking, the drop is accompanied by an increase in the average α-helical tilt angle and a decrease in the lipid order parameter. Inactive proteoliposomes reconstituted at low lipid-to-protein ratios regain full counterflow activity after supplementation with phospholipid to a ratio of about 800:1 (mol/mol) and sonication (data not shown). Thus reconstitution at low lipid-to-protein ratios does not denature the permease. No significant effect of substrate, TDG, or lactose is observed with respect to secondary structure content or helical tilt.
Figure 5.

Dependence of lipid order parameter, the average α-helical tilt angle and counterflow activity on the lipid-to-protein ratio of the sample. For determination of the lipid order parameter and the average α-helical tilt angles, the symmetric methylene stretching modes, and the amide I modes, respectively, were analyzed. All three independent measures reach a plateau in a bilayer lipid environment of ≈800 lipid molecules per permease molecule. For counterflow, the initial rates of [1-14C]lactose accumulation for each lipid-to-protein ratio are shown.
DISCUSSION
In this work, the secondary structure of lactose permease and its orientation in the bilayer were determined by polarized ATR-FTIR spectroscopy. The average α-helical tilt angle of the protein, counterflow activity, and the lipid order parameter are clearly dependent on the lipid-to-protein ratio.
To ensure activity of the protein samples used for FTIR spectroscopy, counterflow assays were performed. Strikingly, the activity of the rehydrated samples is comparable to the activity of control proteoliposomes. With a helical content of 65%, this study confirms the overall secondary structure of the permease, as predicted by numerous other approaches (1). The occurrence of β-sheet is in agreement with earlier observations by Raman spectroscopy (17). Recent EPR studies suggest that the epitope for monoclonal antibody 4B1, which is located in loop VII/VIII, may be β-sheet (J. Voss, W. L. Hubbell, and H.R.K., unpublished data).
The unusually high rate and extent of H/D exchange (90–95%) reflects an unexpected accessibility of the protein backbone to bulk water which is also observed for the human erythrocyte glucose transporter (15). Furthermore, there is a large amount of bound water in the dried protein. Finally, engineered bis- or tris-His residues buried between certain transmembrane helices bind Mn2+, and the pKa for metal binding is similar to that of unperturbed imidazole (19). This high degree of hydration suggests a common feature for transporters of hydrophilic substrates. In contrast, other membrane proteins such as EmrE, a multidrug transporter for hydrophobic substrates, or bacteriorhodopsin, a light-driven proton pump, exhibit much less amide H/D exchange (20, 21).
In this study, no substrate effect on secondary structure or average helical tilt angle in the membrane is observed, even though widespread conformational changes upon ligand binding have been reported (1, 22). Two explanations for this seeming contradiction are plausible: (i), it is possible that either the presence of a proton electrochemical gradient or a sugar gradient across the membrane are required to induce conformational changes in the lipid bilayer, and (ii) crosslinking experiments indicate that substrate binding induces rigid body displacement of entire transmembrane α-helices (23).
Upon increasing the lipid-to-protein ratio (i.e., dilution of the permease in the membrane), a decrease in the average helical tilt angle is observed that correlates with an increase in activity. An important influence of the lipid environment on helix packing of the permease has been observed in previous studies (24). Thus, excimer flourescence between helices is abolished in the presence of sodium dodecylsulfate, whereas an excimer within a single transmembrane helix is retained. The data presented here indicate that the amount of unperturbed bilayer is crucial for proper structure and function of the permease, which might imply that a lipid phase transition occurs at increasing protein concentrations. However, the FTIR spectra give no indication for such a transition (i.e., no change in the crystal field splitting of the CH2 scissoring vibrational modes or a frequency shift of the CH2 stretching modes is observed) (14). Moreover, Cortijo et al. showed (25) that the abrupt lipid phase transition of a biomembrane smears out and disappears when intrinsic membrane proteins are incorporated. Therefore, a more reasonable explanation for the observations is that PE at a certain lipid-to-protein ratio provides a critical lateral packing pressure (26). Indeed, a study by Lis et al. (27) showed that PE exerts an unusually high lateral packing pressure in the bilayer which was attributed to repulsion in the hydrocarbon region and the head groups, as well as to interfacial tension between the hydrophobic and the hydrophilic layers. Our finding of an increased lipid order parameter upon diluting the protein in the membrane can be understood in the context of an increase in membrane thickness that leads to a decrease in the average helical tilt angle, thereby restoring activity.
On the other hand, protein–protein interactions cannot be excluded as a reason for decreased activity at low lipid-to-protein ratios. However, a rough estimation of the lipid requirement for a single permease molecule on the basis of an average protein diameter of 40 Å (28) and a cross-sectional area of 75 Å2 per membrane lipid head group (27) yields about 25 lipid molecules that can be in direct contact with the permease, thereby limiting the likelihood for protein-protein interactions. Taking this calculation to the extreme, the active permease requires an intact bilayer area with a radius of approximately 100 Å. The isolated permease, a typical 12-helix membrane protein without prosthetic groups or metals, seems to be an extremely flexible, metastable molecule that requires external stabilization by the surrounding membrane. Consequently, the need for a critical lipid environment may help to explain the general difficulty in crystallizing such membrane proteins without lipids.
Acknowledgments
We acknowledge fruitful discussions with Prof. Harden McConnell, (Stanford, University) as well as the outstanding technical assistance of Jenny C. Lee and Sven E. Vahsen. This work was supported in part by National Institutes of Health Grant RO 1 DK51131-01 to H.R.K.. J.leC. is the recipient of a Human Frontier Science Program long-term fellowship and was previously supported by a grant from the Deutsche Forschungsgemeinschaft.
ABBREVIATIONS
- ATR-FTIR
attenuated total reflection Fourier transform infrared
- DM
n-dodecyl β-d-maltopyranoside
- PE
phosphatidylethanolamine (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine)
- PG
phosphatidylglycerol, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- RATR
dichroic ratio
- TDG
β-d-galactopyranosyl-1-thio β-d-galactopyranoside
- Ge
Germanium
References
- 1.Kaback H R. In: Handbook of Biological Physics: Transport Processes in Eukaryotic and Prokaryotic Organisms. Konings W N, Kaback H R, Lolkema J S, editors. Vol. 2. Amsterdam: Elsevier; 1996. pp. 203–227. [Google Scholar]
- 2.Kaback H R, Voss J, Wu J. Curr Opin Struct Biol. 1997;7:537–542. doi: 10.1016/s0959-440x(97)80119-4. [DOI] [PubMed] [Google Scholar]
- 3.Costello M J, Viitanen P, Carrasco N, Foster D L, Kaback H R. J Biol Chem. 1984;259:15579–15586. [PubMed] [Google Scholar]
- 4.Kaback H R. Proc Natl Acad Sci USA. 1997;94:5539–5543. doi: 10.1073/pnas.94.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogdanov M, Sun J, Kaback H R, Dowhan W. J Biol Chem. 1996;271:11615–11618. doi: 10.1074/jbc.271.20.11615. [DOI] [PubMed] [Google Scholar]
- 6.Bogdanov M, Dowhan W. J Biol Chem. 1995;270:732–739. doi: 10.1074/jbc.270.2.732. [DOI] [PubMed] [Google Scholar]
- 7.Seto-Young D, Chen C C, Wilson T H. J Membr Biol. 1985;84:259–267. doi: 10.1007/BF01871389. [DOI] [PubMed] [Google Scholar]
- 8.Viitanen P, Newman M J, Foster D L, Wilson T H, Kaback H R. Methods Enzymol. 1986;125:429–452. doi: 10.1016/s0076-6879(86)25034-x. [DOI] [PubMed] [Google Scholar]
- 9.Harrick N J. Internal Reflection Spectroscopy. Ossining, NY: Harrick Scientific; 1967. [Google Scholar]
- 10.Tsuboi M. J Polymer Sci. 1962;59:139–153. [Google Scholar]
- 11.Krimm S, Bandekar J. Adv Protein Chem. 1986;38:181–364. doi: 10.1016/s0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- 12.Goormaghtigh E, Cabiaux V, Ruysschaert J M. Subcell Biochem. 1994;23:329–362. doi: 10.1007/978-1-4615-1863-1_8. [DOI] [PubMed] [Google Scholar]
- 13.Goormaghtigh E, Cabiaux V, Ruysschaert J M. Subcell Biochem. 1994;23:405–450. doi: 10.1007/978-1-4615-1863-1_10. [DOI] [PubMed] [Google Scholar]
- 14.Lewis R N A H, McElhaney R N. In: Infrared Spectroscopy of Biomolecules. Mantsch H H, Chapman D, editors. New York: Wiley–Liss; 1996. pp. 159–237. [Google Scholar]
- 15.Alvarez J, Lee D C, Baldwin S A, Chapman D. J Biol Chem. 1987;262:3502–3509. [PubMed] [Google Scholar]
- 16.Foster D L, Boublik M, Kaback H R. J Biol Chem. 1983;258:31–34. [PubMed] [Google Scholar]
- 17.Vogel H, Wright J K, Jähnig F. EMBO J. 1985;4:3625–3631. doi: 10.1002/j.1460-2075.1985.tb04126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamm L K, Tatulian S A. Biochemistry. 1993;32:7720–7726. doi: 10.1021/bi00081a017. [DOI] [PubMed] [Google Scholar]
- 19.Jung K, Voss J, He M, Hubbell W L, Kaback H R. Biochemistry. 1995;34:6272–6277. doi: 10.1021/bi00019a003. [DOI] [PubMed] [Google Scholar]
- 20.Downer N W, Bruchman T J, Hazzard J H. J Biol Chem. 1986;261:3640–3647. [PubMed] [Google Scholar]
- 21.Arkin I T, Russ W P, Lebendiker M, Schuldiner S. Biochemistry. 1996;35:7233–7238. doi: 10.1021/bi960094i. [DOI] [PubMed] [Google Scholar]
- 22.Jung H, Jung K, Kaback H R. Protein Sci. 1994;3:1052–1057. doi: 10.1002/pro.5560030707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Kaback H R. J Mol Biol. 1997;270:8–13. doi: 10.1006/jmbi.1997.1099. [DOI] [PubMed] [Google Scholar]
- 24.Jung K, Jung H, Kaback H R. Biochemistry. 1994;33:3980–3985. doi: 10.1021/bi00179a026. [DOI] [PubMed] [Google Scholar]
- 25.Cortijo M, Alonso A, Gomez-Fernandez J C, Chapman D. J Mol Biol. 1982;157:597–618. doi: 10.1016/0022-2836(82)90501-0. [DOI] [PubMed] [Google Scholar]
- 26.de Kruijff B. Nature (London) 1997;386:129–130. doi: 10.1038/386129a0. [DOI] [PubMed] [Google Scholar]
- 27.Lis L J, McAlister M, Fuller N, Rand R P, Parsegian V A. Biophys J. 1982;37:667–672. [PMC free article] [PubMed] [Google Scholar]
- 28.Dornmair K, Corni A F, Wright J K, Jähnig F. EMBO J. 1985;4:3633–3638. doi: 10.1002/j.1460-2075.1985.tb04127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]