

Abstract
By using a protein-design algorithm that quantitatively considers side-chain packing, the effect of specific steric constraints on protein design was assessed in the core of the streptococcal protein G β1 domain. The strength of packing constraints used in the design was varied, resulting in core sequences that reflected differing amounts of packing specificity. The structural flexibility and stability of several of the designed proteins were experimentally determined and showed a trend from well-ordered to highly mobile structures as the degree of packing specificity in the design decreased. This trend both demonstrates that the inclusion of specific packing interactions is necessary for the design of native-like proteins and defines a useful range of packing specificity for the design algorithm. In addition, an analysis of the modeled protein structures suggested that penalizing for exposed hydrophobic surface area can improve design performance.
The placement of hydrophobic amino acids into protein cores is critical for maintaining the highly ordered structures of naturally occurring proteins (1–4). Many designed proteins have been constructed to form a nonpolar core by selecting a suitable pattern of hydrophobic and polar residues (HP pattern) but appear to lack the structural ordering of native proteins (5–7). The omission of specific packing interactions as a design criterion is a possible cause of disorder in designed proteins. In this study, we seek to quantitatively assess both the degree to which specific packing interactions are necessary for the design of well-ordered proteins and the tolerance of native-like structure to variations in core packing patterns.
Previous studies that have examined the role of core packing on protein structure demonstrate that while some variation in the buried positions of a protein is allowed, there are limits on the sequences that result in stable native-like folds (2, 8–11). To generalize these results and to provide a framework to assess designed proteins, we propose the use of an automated side-chain selection algorithm, which explicitly and quantitatively considers specific side-chain packing interactions (12), as the basis of a method to define the need for packing constraints in protein design. Our side-chain selection algorithm screens all possible sequences and finds the optimal amino acid type and side-chain orientation for a given backbone. To correctly account for the torsional flexibility of side chains and the geometric specificity of side-chain placement, we consider a discrete set of all allowed conformers of each side chain, called rotamers (13, 14). The immense search problem presented by rotamer sequence optimization is overcome by application of the dead-end elimination (DEE) theorem (15–17). Our implementation of the DEE theorem extends its utility to sequence design and rapidly finds the globally optimal sequence in its optimal conformation. Scoring of sequence arrangements includes an atomic van der Waals potential that captures the two main features of steric packing interactions: excluded volume and the weakly attractive dispersive force. Protein cores designed with this and with similar (18, 19) algorithms result in stable well-ordered proteins.
The referenced sequence prediction algorithms select a single family of closely related core sequences for a given backbone, indicating that designs produced by these algorithms are highly determined by packing specificity. Two factors are likely to be responsible for this stringency: the use of a fixed backbone and the highly restrictive repulsive (excluded volume) component of the van der Waals potential. The repulsive component can be modulated, however, by scaling the van der Waals radii of the atoms in the simulation. We implement this modulation in the packing constraints by varying a radius scale factor, α (Eq. 1). R0 and D0 are the van der Waals radius and well depth, respectively, and Evdw and R are the energy and interatomic distance.
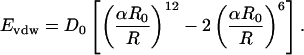 |
1 |
By predicting core sequences with various radii scalings and then experimentally characterizing the resulting proteins, a rigorous study of the importance of packing effects on protein design is possible.
By using a protein design algorithm to assess the bounds of effective steric constraints on core packing, these bounds can be incorporated into the algorithm to improve design performance. Specifically, a reduced van der Waals steric constraint can compensate for the restrictive effect of a fixed backbone and discrete side-chain rotamers in the simulation and could allow a broader sampling of sequences compatible with the desired fold. The use of experimental data to test our designs and subsequently to improve our design algorithm is the central feature of our overall protein design strategy (12). This study should provide practical improvements to our sequence scoring potential in addition to generally assaying the role of packing specificity in protein structure.
METHODS
Sequence Optimization: DEE and Monte Carlo.
The protein structure was modeled on the backbone coordinates of streptococcal protein G β1 domain (Gβ1), Protein Data Bank record 1pga (20, 21). Atoms of all side chains not optimized were left in their crystallographically determined positions. The program biograf (Molecular Simulations, San Diego) was used to generate explicit hydrogens on the structure, which was then conjugate-gradient-minimized for 50 steps using the Dreiding force field (22). The rotamer library, DEE optimization, and Monte Carlo search followed our previous work (12). A Lennard–Jones 12–6 potential was used for van der Waals interactions, with atomic radii scaled for the various cases as discussed in the text. The Richards definition of solvent-accessible surface area (23) was used, and areas were calculated with the Connolly algorithm (24). An atomic solvation parameter, derived from our previous work, of 23 cal per mol per Å2 (1 cal = 4.184 J) was used to favor hydrophobic burial and to penalize solvent exposure. To calculate side-chain nonpolar exposure in our optimization framework, we first consider the total hydrophobic area exposed by a rotamer in isolation. This exposure is decreased by the area buried in rotamer/template contacts, and the sum of the areas buried in pairwise rotamer/rotamer contacts.
Peptide Synthesis and Purification.
With the exception of the 11 core positions designed by the sequence selection algorithm, the sequences synthesized match Protein Data Bank entry 1pga. Peptides were synthesized by using standard fluorenylmethoxycarbonyl chemistry, and were purified by reverse-phase HPLC. Matrix-assisted laser desorption mass spectrometry found all molecular weights to be within one unit of the expected masses.
CD and Fluorescence Spectroscopy and Size-Exclusion Chromatography.
The solution conditions for all experiments were 50 mM sodium phosphate buffer at pH 5.5 and 25°C unless noted. CD spectra were acquired on an Aviv 62DS spectrometer equipped with a thermoelectric unit. Peptide concentration was approximately 20 μM. Thermal melts were monitored at 218 nm by using 2° increments with an equilibration time of 120 s. Melting temperature (Tm) was defined as the maximum of the derivative of the melting curve. Reversibility for each of the proteins was confirmed by comparing room temperature CD spectra from before and after heating. Guanidinium chloride denaturation measurements followed published methods (25). Protein concentrations were determined by UV spectrophotometry. Fluorescence experiments were performed on a Hitachi F-4500 in a 1-cm-pathlength cell. Both peptide and 8-anilino-1-naphthalene sulfonic acid (ANS) concentrations were 50 μM. The excitation wavelength was 370 nm and emission was monitored from 400 to 600 nm. Size-exclusion chromatography was performed with a PolyLC hydroxyethyl A column at pH 5.5 in 50 mM sodium phosphate at 0°C. Ribonuclease A, carbonic anhydrase, and Gβ1 were used as molecular weight standards. Peptide concentrations during the separation were ∼15 μM, as estimated from peak heights monitored at 275 nm.
NMR Spectroscopy.
Samples were prepared in 90:10 H2O/2H2O and 50 mM sodium phosphate buffer at pH 5.5. Spectra were acquired on a Varian Unityplus 600-MHz spectrometer at 25°C. Samples were approximately 1 mM, except for α70, which had limited solubility (100 μM). For hydrogen exchange studies, an NMR sample was prepared, the pH was adjusted to 5.5, and a spectrum was acquired to serve as an unexchanged reference. This sample was lyophilized, reconstituted in 2H2O, and repetitive acquisition of spectra was begun immediately at a rate of 75 s per spectrum. Data acquisition continued for ∼20 h, and then the sample was heated to 99°C for 3 min to fully exchange all protons. After cooling to 25°C, a final spectrum was acquired to serve as the fully exchanged reference. The areas of all exchangeable amide peaks were normalized by a set of nonexchanging aliphatic peaks. pH values, uncorrected for isotope effects, were measured for all the samples after data acquisition, and the time axis was normalized to correct for minor differences in pH (26).
RESULTS
Model System Core Sequence Predictions.
An ideal model system to study core packing is Gβ1 (20, 27–31). Its small size, 56 residues, renders computations and experiments tractable. Perhaps most critical for a core packing study, Gβ1 contains no disulfide bonds and does not require a cofactor or metal ion to fold. Further, Gβ1 contains sheet, helix, and turn structures and is without the repetitive side-chain packing patterns found in coiled coils or some helical bundles. This lack of periodicity reduces the bias from a particular secondary or tertiary structure and necessitates the use of an objective side-chain selection algorithm to examine packing effects.
Sequence positions that constitute the core were chosen by examining the side-chain solvent-accessible surface area of Gβ1. Any side chain exposing less than 10% of its surface was considered buried. Eleven residues meet this criteria, with 7 from the β-sheet (positions 3, 5, 7, 20, 43, 52, and 54), three from the helix (positions 26, 30, and 34) and 1 in an irregular secondary structure (position 39). These positions form a contiguous core. The remainder of the protein structure, including all other side chains and the backbone, was used as the template for sequence selection calculations at the 11 core positions.
All possible core sequences consisting of alanine, valine, leucine, isoleucine, phenylalanine, tyrosine, or tryptophan were considered. Our rotamer library was similar to that used by Desmet et al. (15). Optimizing the sequence of the core of Gβ1 with 217 possible hydrophobic rotamers at all 11 positions results in 21711 or 5 × 1025 rotamer sequences. Our scoring function consisted of two components: a van der Waals energy term and an atomic solvation term favoring burial of hydrophobic surface area. The van der Waals radii of all atoms in the simulation were scaled by a factor α (Eq. 1) to change the importance of packing effects. Radii were not scaled for the buried surface area calculations. Global optimum sequences for various values of the radius scaling factor α were found using the DEE theorem (Table 1). Optimal sequences, and their corresponding proteins, are named by the radius scale factor used in their design. For example, the sequence designed with a radius scale factor of α = 0.90 is called α90.
Table 1.
Optimal core sequences and relative side-chain volume vs. α
| α | Vol | Gβ1 sequence
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TYR-3 | LEU-5 | LEU-7 | ALA-20 | ALA-26 | PHE-30 | ALA-34 | VAL-39 | TRP-43 | PHE-52 | VAL-54 | ||
| 0.70 | 1.28 | TRP | TYR | ILE | ILE | PHE | TRP | LEU | ILE | PHE | LEU | ILE |
| 0.75 | 1.23 | PHE | ILE | PHE | ILE | VAL | TRP | VAL | LEU | | | | | ILE |
| 0.80 | 1.13 | PHE | | | ILE | | | | | | | ILE | ILE | | | TRP | ILE |
| 0.85 | 1.15 | PHE | | | ILE | | | | | | | LEU | ILE | | | TRP | PHE |
| 0.90 | 1.01 | PHE | | | ILE | | | | | | | | | ILE | | | | | | |
| 0.95 | 1.01 | PHE | | | ILE | | | | | | | | | ILE | | | | | | |
| 1.0 | 0.99 | PHE | | | VAL | | | | | | | | | ILE | | | | | | |
| 1.05 | 0.93 | PHE | | | ALA | | | | | | | | | | | | | | | | |
| 1.075 | 0.83 | ALA | ALA | ILE | | | | | ILE | | | | | | | ILE | ILE |
| 1.10 | 0.77 | ALA | | | ALA | | | | | ALA | | | | | | | ILE | ILE |
| 1.15 | 0.68 | ALA | ALA | ALA | | | | | ALA | | | | | | | LEU | | |
α100 was designed with α = 1.0 and hence serves as a baseline for full incorporation of steric effects. The α100 sequence is very similar to the core sequence of Gβ1 (Table 1) even though no information about the naturally occurring sequence was used in the side-chain selection algorithm. Variation of α from 0.90 to 1.05 caused little change in the optimal sequence, demonstrating the algorithm’s robustness to minor parameter perturbations. Further, the packing arrangements predicted with α = 0.90 to 1.05 closely match Gβ1 with average χ angle differences of only 4° from the crystal structure. The high identity and conformational similarity to Gβ1 imply that, when packing constraints are used, backbone conformation strongly determines a single family of well-packed core designs. Nevertheless, the constraints on core packing were being modulated by α as demonstrated by Monte Carlo searches for other low-energy sequences. Several alternate sequences and packing arrangements are in the 20 best sequences found by the Monte Carlo procedure when α = 0.90. These alternate sequences score much worse when α = 0.95, and when α = 1.0 or 1.05, only strictly conservative packing geometries have low energies. Therefore, α = 1.05 and α = 0.90 define the high and low ends, respectively, of a range where packing specificity dominates sequence design.
For α < 0.90, the role of packing is reduced enough to let the hydrophobic surface potential begin to dominate, thereby increasing the size of the residues selected for the core (Table 1). A significant change in the optimal sequence appears between α = 0.90 and 0.85 with both α85 and α80 containing three additional mutations relative to α90. Also, α85 and α80 have a 15% increase in total side-chain volume relative to Gβ1. As α drops below 0.80, an additional 10% increase in side-chain volume and numerous mutations occur, showing that packing constraints have been overwhelmed by the drive to bury nonpolar surface. Though the jumps in volume and shifts in packing arrangement appear to occur suddenly for the optimal sequences, examination of the suboptimal low-energy sequences by Monte Carlo sampling demonstrates that the changes are not abrupt. For example, the α85 optimal sequence is the 11th best sequence when α = 0.90, and similarly, the α90 optimal sequence is the 9th best sequence when α = 0.85.
For α > 1.05, atomic van der Waals repulsions are so severe that most amino acids cannot find any allowed packing arrangements, resulting in the selection of alanine for many positions. This stringency is likely an artifact of the large atomic radii and does not reflect increased packing specificity accurately. Rather, α = 1.05 is the upper limit for the usable range of van der Waals scales within our modeling framework.
Experimental Characterization of Core Designs.
Variation of the van der Waals scale factor α results in four regimes of packing specificity: regime 1, where 0.9 ≤ α ≤ 1.05 and packing constraints dominate the sequence selection; regime 2, where 0.8 ≤ α < 0.9 and the hydrophobic solvation potential begins to compete with packing forces; regime 3, where α < 0.8 and hydrophobic solvation dominates the design; regime 4, where α > 1.05 and van der Waals repulsions appear to be too severe to allow meaningful sequence selection. Sequences that are optimal designs were selected from each of the regimes for synthesis and characterization. They are α90 from regime 1, α85 from regime 2, α70 from regime 3, and α107 from regime 4. For each of these sequences, the calculated amino acid identities of the 11 core positions are shown in Table 1; the remainder of the protein sequence matches Gβ1. The goal was to study the relation between the degree of packing specificity used in the core design and the extent of native-like character in the resulting proteins.
α90 and α85 have ellipticities and spectra very similar to Gβ1 (data not shown), suggesting that their secondary structure content is comparable to that of Gβ1 (Fig. 1A). Conversely, α70 has much weaker ellipticity and a perturbed spectrum, implying a loss of secondary structure relative to Gβ1. α107 has a spectrum characteristic of a random coil. Thermal melts monitored by CD are shown in Fig. 1B. α85 and α90 both have cooperative transitions with Tm values of 83°C and 92°C, respectively. α107 shows no thermal transition, behavior expected from a fully unfolded polypeptide, and α70 has a broad shallow transition, centered at ∼40°C, characteristic of partially folded structures. Relative to Gβ1, which has a Tm of 87°C (28), α85 is slightly less thermostable and α90 is more stable. Chemical denaturation measurements of the free energy of unfolding (ΔGu) at 25°C match the trend in the Tm. α90 has a larger ΔGu than that reported for Gβ1 (28) whereas α85 is slightly less stable. It was not possible to measure ΔGu for α70 or α107 because they lack discernible transitions.
Figure 1.

Secondary structure and thermal stability of α90, α85, α70, and α107. (A) Far-UV CD spectra. (B) Thermal denaturation monitored by CD.
The extent of chemical shift dispersion in the proton NMR spectrum of each protein was assessed to gauge each protein’s degree of native-like character (Fig. 2). α90 possesses a highly dispersed spectrum, the hallmark of a well-ordered native protein. α85 has diminished chemical-shift dispersion and peaks that are somewhat broadened relative to α90, suggesting a moderately mobile structure that nevertheless maintains a distinct fold. α70’s NMR spectrum has almost no dispersion. The broad peaks are indicative of a collapsed but disordered and fluctuating structure. α107 has a spectrum with sharp lines and no dispersion, which is indicative of an unfolded protein.
Figure 2.

Proton NMR spectra of α90, α85, α70, α107, and α85W43V. The decrease in dispersion from α90 to α85 to α70 reflects a graded decrease in protein structural order. α107 appears unfolded. α85W43V has narrower lines and greater dispersion than α85, indicating that the single Trp → Val mutation reduced conformational flexibility. The sharp peaks at 8.45 and 0.15 ppm in the α70 spectrum are impurities.
Amide hydrogen exchange kinetics are consistent with the conclusions reached from examination of the proton NMR spectra. Fig. 3 shows the average number of unexchanged amide protons as a function of time for each of the designed proteins. α90 protects ∼13 protons for more than 20 h of exchange at pH 5.5 and 25°C. The α90 exchange curve is indistinguishable from that of Gβ1 (data not shown). α85 also maintains a well-protected set of amide protons, a distinctive feature of ordered native-like proteins. The number of protected protons, however, is only about half that of α90. The difference is likely due to higher flexibility in some parts of the α85 structure. In contrast, α70 and α107 were fully exchanged within the 3-min dead time of the experiment, indicating highly dynamic structures.
Figure 3.

Amide hydrogen–deuterium exchange kinetics of α90, α85, α70, α107, and α85W43V. Total area of exchangeable peaks, expressed as number of protons, as a function of exchange time at 25°C and pH 5.5.
Near UV CD spectra and the extent of ANS binding were used to assess the structural ordering of the proteins. The near-UV CD spectra of α85 and α90 have strong peaks, as expected for proteins with aromatic residues fixed in a unique tertiary structure whereas α70 and α107 have featureless spectra indicative of proteins with mobile aromatic residues, such as nonnative collapsed states or unfolded proteins. α70 also binds ANS well, as indicated by a 3-fold intensity increase and blue shift of the ANS emission spectrum. This strong binding suggests that α70 possesses a loosely packed or partially exposured cluster of hydrophobic residues accessible to ANS. ANS binds α85 weakly, with only a 25% increase in emission intensity, similar to the association seen for some native proteins (32). α90 and α107 cause no change in ANS fluorescence. All of the proteins migrated as monomers during size-exclusion chromatography.
DISCUSSION
In summary, α90 is a well-packed native-like protein by all criteria, and it is more stable than the naturally occurring Gβ1 sequence, possibly because of increased hydrophobic surface burial. α85 is also a stable ordered protein, albeit with greater motional flexibility than α90, as shown by its NMR spectrum and hydrogen-exchange behavior. α70 has all the features of a disordered collapsed globule: a noncooperative thermal transition, no NMR spectral dispersion or amide proton protection, reduced secondary structure content, and strong ANS binding. α107 is a completely unfolded chain, likely due to its lack of large hydrophobic residues to hold the core together. The clear trend is a loss of protein ordering as α decreases below 0.90.
The different packing regimes for protein design can be evaluated in light of the experimental data. In regime 1, with 0.9 ≤ α ≤ 1.05, the design is dominated by packing specificity resulting in well-ordered proteins. In regime 2, with 0.8 ≤ α < 0.9, packing forces are weakened enough to let the hydrophobic force drive larger residues into the core, which produces a stable well-packed protein with somewhat increased structural motion. In regime 3, α < 0.8, packing forces are reduced to such an extent that the hydrophobic force dominates, resulting in a fluctuating, partially folded structure with no stable core packing. In regime 4, α > 1.05, the steric forces used to implement packing specificity are scaled too high to allow reasonable sequence selection and hence produce an unfolded protein. These results indicate that effective protein design requires a consideration of packing effects. Within the context of a protein design algorithm, we have quantitatively defined the range of packing forces necessary for successful designs.
To take advantage of the benefits of reduced packing constraints, protein cores should be designed with the smallest α that still results in structurally ordered proteins. The optimal protein sequence from regime 2, α85, is stable and well packed, suggesting 0.8 ≤ α < 0.9 as a good range. NMR spectra and hydrogen-exchange kinetics, however, clearly show that α85 is not as structurally ordered as α90. The packing arrangements predicted by our algorithm for Trp-43 in α85 and α90 present a possible explanation (Fig. 4). For α90, Trp-43 is predicted to pack in the core with the same conformation as in the crystal structure of Gβ1. In α85, the larger side chains at positions 34 and 54, leucine and phenylalanine, respectively, compared with alanine and valine in α90, force Trp-43 to expose 91 Å2 of nonpolar surface compared with 19 Å2 in α90. The hydrophobic driving force this exposure represents seems likely to stabilize alternate conformations that bury Trp-43 and thereby could contribute to α85’s conformational flexibility (34, 35). In contrast to the other core positions, a residue at position 43 can be mostly exposed or mostly buried depending on its side-chain conformation. We designate positions with this characteristic as boundary positions, which pose a difficult problem for protein design because of their potential to either strongly interact with the protein’s core or with solvent.
Figure 4.

Core packing arrangements predicted by DEE for α90 (Upper) and α85 (Lower). Only side chains for residues 34, 39, 43, 52, and 54 are shown. In α90, Trp-43 buries more than 90% of its surface area. In α85, Trp-43 is only 46% buried and is rotated into solvent to avoid steric clashes with Leu-34 and Phe-52, which occupy a larger volume than Ala-34 and Val-52 in α90. Figures were produced with molmol (33).
A scoring function that penalizes the exposure of hydrophobic surface area might assist in the design of boundary residues. Dill and coworkers (36) used an exposure penalty to improve protein designs in a theoretical study. A nonpolar exposure penalty would favor packing arrangements that either bury large side chains in the core or replace the exposed amino acid with a smaller or more polar one. We implemented a side-chain nonpolar exposure penalty in our optimization framework and used a penalizing solvation parameter with the same magnitude as the hydrophobic burial parameter.
The results of adding a hydrophobic surface exposure penalty to our scoring function are shown in Table 2. When α = 0.85, the nonpolar exposure penalty dramatically alters the ordering of low-energy sequences. The α85 sequence, the former ground state, drops to 7th and the rest of the 15 best sequences expose far-less hydrophobic area because they bury Trp-43 in a conformation similar to α90 (Fig. 4). The exceptions are the 8th and 14th sequences, which reduce the size of the exposed boundary residue by replacing Trp-43 with an isoleucine, and the 13th best sequence, which replaces Trp-43 with a valine. The new ground-state sequence is very similar to α90, with a single valine → isoleucine mutation, and should share α90’s stability and structural order. In contrast, when α = 0.90, the optimal sequence does not change and the next 14 best sequences, found by Monte Carlo sampling, change very little. This minor effect is not surprising, since steric forces still dominate for α = 0.90 and most of these sequences expose very little surface area. Burying Trp-43 restricts sequence selection in the core somewhat, but the reduced packing forces for α = 0.85 still produce more sequence variety than α = 0.90. The exposure penalty complements the use of reduced packing specificity by limiting the gross overpacking and solvent exposure that occurs when the core’s boundary is disrupted. Adding this constraint should allow lower packing forces to be used in protein design, resulting in a broader range of high-scoring sequences and reduced bias from fixed backbone and discrete rotamers.
Table 2.
Exposure penalty effect on sequence selection and exposed surface area (Anp)
| No. | Anp | TYR-3 | LEU-5 | LEU-7 | ALA-20 | ALA-26 | PHE-30 | ALA-34 | VAL-39 | TRP-43 | PHE-52 | VAL-54 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. α = 0.85 | ||||||||||||
| 1 | 109 | PHE | | | ILE | | | | | | | LEU | ILE | | | TRP | PHE |
| 2 | 109 | | | | | ILE | | | | | | | LEU | ILE | | | TRP | PHE |
| 3 | 104 | PHE | | | ILE | | | | | | | LEU | ILE | | | | | PHE |
| 4 | 104 | | | | | ILE | | | | | | | LEU | ILE | | | | | PHE |
| 5 | 108 | PHE | | | ILE | | | | | | | LEU | | | | | TRP | PHE |
| 6 | 62 | PHE | | | ILE | | | | | | | LEU | ILE | VAL | TRP | PHE |
| 7 | 103 | PHE | | | ILE | | | | | | | LEU | ILE | | | TYR | PHE |
| 8 | 109 | PHE | | | VAL | | | | | | | LEU | ILE | | | TRP | PHE |
| 9 | 30 | PHE | | | ILE | | | | | | | | | ILE | | | | | | |
| 10 | 38 | PHE | | | ILE | | | | | | | | | ILE | | | TRP | | |
| 11 | 108 | | | | | ILE | | | | | | | LEU | | | | | TRP | PHE |
| 12 | 62 | | | | | ILE | | | | | | | LEU | ILE | VAL | TRP | PHE |
| 13 | 109 | PHE | | | ILE | | | | | TYR | LEU | ILE | | | TRP | PHE |
| 14 | 103 | | | | | ILE | | | | | | | LEU | ILE | | | TYR | PHE |
| 15 | 109 | | | | | VAL | | | | | | | LEU | ILE | | | TRP | PHE |
| B. α = 0.85 exposure penalty | ||||||||||||
| 1 | 30 | PHE | | | ILE | | | | | | | | | ILE | | | | | ILE |
| 2 | 29 | PHE | | | ILE | | | | | | | ILE | ILE | | | | | | |
| 3 | 29 | PHE | ILE | PHE | | | | | | | | | ILE | | | | | | |
| 4 | 30 | | | | | ILE | | | | | | | | | ILE | | | | | ILE |
| 5 | 29 | | | | | ILE | | | | | | | ILE | ILE | | | | | | |
| 6 | 29 | | | ILE | PHE | | | | | | | | | ILE | | | | | | |
| 7 | 109 | PHE | | | ILE | | | | | | | LEU | ILE | | | TRP | PHE |
| 8 | 52 | PHE | | | ILE | | | | | | | LEU | ILE | ILE | | | PHE |
| 9 | 29 | | | | | ILE | | | | | | | | | ILE | | | | | | |
| 10 | 29 | PHE | | | ILE | | | | | | | | | ILE | | | | | | |
| 11 | 109 | | | | | ILE | | | | | | | LEU | ILE | | | TRP | PHE |
| 12 | 38 | PHE | | | ILE | | | | | | | | | ILE | | | TRP | ILE |
| 13 | 62 | PHE | | | ILE | | | | | | | LEU | ILE | VAL | TRP | PHE |
| 14 | 52 | | | | | ILE | | | | | | | LEU | ILE | ILE | | | PHE |
| 15 | 30 | PHE | | | ILE | | | | | | | | | ILE | | | TYR | ILE |
To examine the effect of substituting a smaller residue at a boundary position, we synthesized and characterized the 13th best sequence of the α = 0.85 optimization with exposure penalty (Table 2, section B). This sequence, α85W43V, replaces Trp-43 with a valine but is otherwise identical to α85. Though the 8th and 14th sequences also have a smaller side chain at position 43, additional changes in their sequences relative to α85 would complicate interpretation of the effect of the boundary position change. Also, α85W43V has a significantly different packing arrangement compared with Gβ1, with 7 out of 11 positions altered, but only an 8% increase in side-chain volume. Hence, α85W43V is a test of the tolerance of this fold to a different, but nearly volume-conserving, core. The far UV CD spectrum of α85W43V is very similar to that of Gβ1 with an ellipticity at 218 nm of −14000 deg⋅cm2/dmol. While the secondary structure content of α85W43V is native-like, its Tm is 65°C, nearly 20°C lower than α85. In contrast to α85W43V’s decreased stability, its NMR spectrum has greater chemical shift dispersion than α85 (Fig. 2). The amide hydrogen-exchange kinetics show a well-protected set of about four protons after 20 h (Fig. 3). This faster exchange relative to α85 is explained by α85W43V’s significantly lower stability (37). α85W43V appears to have improved structural specificity at the expense of stability, a phenomenon observed previously in coiled coils (38). By using an exposure penalty, the design algorithm produced a protein with greater native-like character.
We have quantitatively defined the role of packing specificity in protein design and have provided practical bounds for the role of steric forces in our protein design algorithm. This study differs from previous work because of the use of an objective quantitative algorithm to vary packing forces during design. Further, by using the minimum effective level of steric forces, we were able to design a wider variety of packing arrangements that were compatible with the Gβ1 fold. Finally, we have identified a difficulty in the design of side chains that lie at the boundary between the core and the surface of the protein, and we have implemented a nonpolar surface exposure penalty in our sequence design scoring function that addresses this problem.
Acknowledgments
We thank D. B. Gordon for helpful discussions, S. Ross for assistance with the NMR spectroscopy, and G. Hathaway for mass spectra. This work was supported by the Rita Allen Foundation, the David and Lucile Packard Foundation, and the Searle Scholars Program/The Chicago Community Trust. B.I.D. is partially supported by National Institutes of Health Training Grant GM 08346.
ABBREVIATIONS
- DEE
dead-end elimination
- Tm
melting temperature
- Gβ1
streptococcal protein G β1 domain
- ANS
8-anilino-1-naphthalene sulfonic acid
References
- 1.Shortle D, Stites W, Meeker A. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 2.Lim W A, Sauer R T. J Mol Biol. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- 3.Richards F M, Lim W A. Q Rev Biophys. 1993;26:423–498. doi: 10.1017/s0033583500002845. [DOI] [PubMed] [Google Scholar]
- 4.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regan L, DeGrado W F. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 6.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 7.Kamtekar S, Schiffer J M, Xiong H, Babik J M, Hecht M H. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 8.Lim W A, Sauer R T. Nature (London) 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 9.Lim W A, Farruggio D C, Sauer R T. Biochemistry. 1992;31:4324–4333. doi: 10.1021/bi00132a025. [DOI] [PubMed] [Google Scholar]
- 10.Munson M, O’Brien R, Sturtevant J M, Regan L. Protein Sci. 1994;3:2015–2022. doi: 10.1002/pro.5560031114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munson M, Balasubramanian S, Fleming K G, Nagi A D, O’Brien R, Sturtevant J M, Regan L. Protein Sci. 1996;5:1584–1593. doi: 10.1002/pro.5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahiyat B I, Mayo S L. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 14.Dunbrack R L, Karplus M. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 15.Desmet J, De Maeyer M, Hazes B, Lasters I. Nature (London) 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 16.Desmet J, De Maeyer M, Lasters I. In: The Dead-End Elimination Theorem: A New Approach To The Side-Chain Packing Problem. Merz K Jr, Le Grand S, editors. Boston: Birkhauser; 1994. pp. 307–337. [Google Scholar]
- 17.Goldstein R F. Biophys J. 1994;66:1335–1340. doi: 10.1016/S0006-3495(94)80923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desjarlais J R, Handel T M. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Betz S F, Degrado W F. Biochemistry. 1996;35:6955–6962. doi: 10.1021/bi960095a. [DOI] [PubMed] [Google Scholar]
- 20.Gallagher T, Alexander P, Bryan P, Gilliland G L. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
- 21.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 22.Mayo S L, Olafson B D, Goddard W A., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 23.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 24.Connolly M L. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 25.Pace C N. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 26.Rohl C A, Scholtz J M, York E J, Stewart J M, Baldwin R L. Biochemistry. 1992;31:1263–1269. doi: 10.1021/bi00120a001. [DOI] [PubMed] [Google Scholar]
- 27.Gronenborn A M, Filpula D R, Essig N Z, Achari A, Whitlow M, Wingfield P T, Clore G M. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 28.Alexander P, Fahnestock S, Lee T, Orban J, Bryan P. Biochemistry. 1992;31:3597–3603. doi: 10.1021/bi00129a007. [DOI] [PubMed] [Google Scholar]
- 29.Barchi J J, Grasberger B, Gronenborn A M, Clore G M. Protein Sci. 1994;3:15–21. doi: 10.1002/pro.5560030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuszewski J, Clore G M, Gronenborn A M. Protein Sci. 1994;3:1945–1952. doi: 10.1002/pro.5560031106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orban J, Alexander P, Bryan P, Khare D. Biochemistry. 1995;34:15291–15300. doi: 10.1021/bi00046a038. [DOI] [PubMed] [Google Scholar]
- 32.Semisotnov G V, Rodionova N A, Razgulyaev O I, Uversky V N, Gripas A F, Gilmanshin R I. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 33.Koradi R, Billeter M, Wuthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 34.Dill K A. Biochemistry. 1985;24:1501–1509. doi: 10.1021/bi00327a032. [DOI] [PubMed] [Google Scholar]
- 35.Onuchic J N, Socci N D, Lutheyschulten Z, Wolynes P G. Folding and Design. 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Brem R, Chan H S, Dill K A. Protein Eng. 1995;8:1205–1213. doi: 10.1093/protein/8.12.1205. [DOI] [PubMed] [Google Scholar]
- 37.Mayo S L, Baldwin R L. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 38.Harbury P B, Zhang T, Kim P S, Alber T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]