

Crystals of an MHC class I molecule bound to naturally occurring peptide variants from the dengue virus NS3 protein contained high levels of solvent and required optimization of cryoprotectant and dehydration protocols for each complex to yield well ordered diffraction, a process facilitated by the use of a free-mounting system.
Keywords: MHC class I, free-mounting system, crystal dehydration
Abstract
T-cell recognition of the antigenic peptides presented by MHC class I molecules normally triggers protective immune responses, but can result in immune enhancement of disease. Cross-reactive T-cell responses may underlie immunopathology in dengue haemorrhagic fever. To analyze these effects at the molecular level, the functional MHC class I molecule HLA-A*1101 was crystallized bound to six naturally occurring peptide variants from the dengue virus NS3 protein. The crystals contained high levels of solvent and required optimization of the cryoprotectant and dehydration protocols for each complex to yield well ordered diffraction, a process that was facilitated by the use of a free-mounting system.
1. Introduction
Dengue is an emerging viral disease that is now responsible for frequent epidemics in a large number of tropical and subtropical countries (Gubler, 1998 ▶). Dengue virus is an arthropod-borne flavivirus that can cause haemorrhagic fever (World Health Organization, 2002 ▶). There are four serotypes of the virus and immunity to one serotype does not provide protection against the others (World Health Organization, 2002 ▶). Indeed, there is evidence that immune enhancement of disease may be a consequence of cross-reactive T-cell responses (Mongkolsapaya et al., 2003 ▶, 2006 ▶). The four dengue virus serotypes differ by up to 30% in amino-acid sequence and are almost as divergent as dengue virus is from other members of the flavivirus family. Since immunity to one serotype does not confer protection to other serotypes, serial infections are common and these can lead to life-threatening haemorrhagic fever. Immune enhancement is implicated since severe cases occur in individuals who have previously encountered the virus (Guzman et al., 2000 ▶).
The T-cell response in dengue has been characterized in detail and we have described responses to an epitope (termed GTS in accordance with the single-letter code for the first three residues of the epitope) in nonstructural protein 3 (NS3), which has six different sequence variants among the dengue-virus serotypes (Mongkolsapaya et al., 2003 ▶). This epitope is presented in the form of a 10-mer peptide complexed by the MHC class I molecule HLA-A*1101 (HLA-A11). A variety of T-cell responses can be made as a result of T-cell receptor recognition of these peptide–MHC class I (pMHC) complexes; some T cells are highly specific to a single variant, whilst other T cells can be highly promiscuous. In order to understand the molecular basis of this specificity and cross-reactivity, we set out to solve the structure of HLA-A11 in complex with each of the six peptides. The six pMHC complexes crystallized under closely related conditions which, in their absence of PEG-type precipitants, differed markedly from those typically reported for pMHC complexes in general and peptide–HLA-A11 complexes in particular. Bragg diffraction to sufficient resolution for structure determination of the full set of complexes was only obtained after extensive optimization of the cryoprotectant and dehydration procedures. Possibly as a result of their high solvent content (∼70%), these crystals therefore provide a clear example of the utility of humidity-controlled free-mounting techniques.
2. Materials and methods
2.1. Protein expression and purification
Soluble pMHC samples of all six variant peptides (termed GTS1.1, GTS2.1, GTS2.2, GTS2.3, GTS2.4 and GTS3.1 as defined in Table 1 ▶) in complex with HLA-A*1101 (HLA-A11) were produced using previously described methods (Reid et al., 1996 ▶). Briefly, the extracellular region of the HLA-A11 heavy chain (residues 1–275; with and without the BirA-recognizing sequence) and β2 microglobulin (β2m) were expressed separately in Escherichia coli strain BL21 using the pET23d+ and pGMT7 expression systems, respectively, and isopropyl β-d-thiogalactosidase (Melford, UK) as an inducer. The peptides were synthesized in-house on a 396 MPS automated peptide synthesizer (Advanced ChemTech) and their purity was characterized by reverse-phase HPLC (Gilson).
Table 1. Dengue-peptide variants.
| Variant | Dengue serotype | Sequence |
|---|---|---|
| GTS1.1 | Den 1 | GTSGSPIVNR |
| GTS2.1 | Den 2 | GTSGSPIIDK |
| GTS2.2 | Den 2 | GTSGSPIVDR |
| GTS2.3 | Den 2 | GTSGSPIVDK |
| GTS2.4 | Den 2 | GTSGSPIADK |
| GTS3.1 | Den 3 or Den 4 | GTSGSPIINR |
Inclusion bodies of HLA-A11 heavy chain and β2m were washed (in 0.5% Triton X-100, 50 mM Tris pH 8.0, 100 mM NaCl, 0.1% sodium azide and 2 mM DTT), solubilized in urea (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris pH 8.0, 0.1 M EDTA and 0.1 mM DTT) and refolded in the presence of peptide at a molar ratio of 2:5:10 heavy chain:β2m:peptide at 277 K for 48 h using a refolding buffer (100 mM Tris pH 8.0, 400 mM l-arginine, 2 mM EDTA pH 8.0, 5 mM reduced glutathione, 0.5 mM oxidized glutathione and 100 µM PMSF protease inhibitor). pMHC complexes with the BirA-recognizing sequence were biotinylated using the BirA enzyme (Avidity, Denver, CO, USA) following the company protocol. Biotinylated and nonbiotinylated samples were purified by FPLC size-exclusion gel filtration on a Sephadex 75 26/60 column (Pharmacia, UK) using 20 mM Tris pH 8.0 and 25 mM NaCl buffer, further purified on a 1 ml HiTrap Q HP anion-exchange column (Pharmacia, UK) and kept at 193 K until required. The nonbiotinylated pMHC complexes were buffer-exchanged to 7 mM Tris pH 7.0 and concentrated to 10–12 mg ml−1 prior to crystallization. Phycoerythrin-labelled pMHC tetrameric complexes (PE-tetramers) were prepared by mixing biotinylated pMHC complexes with fluorochrome-conjugated streptavidin as described previously (Altman et al., 1996 ▶).
2.2. Conformational and functional analysis of pMHC complexes
The refolded pMHCs were tested for correct folding by dot enzyme-linked immunoassay (DEIA) using the monoclonal antibody W6/32 (which recognizes a conformational epitope of a pMHC complex; Shields & Ribaudo, 1998 ▶) and their functionality was verified using a competitive binding assay as described previously (Mongkolsapaya et al. 2003 ▶) with slight modification. GTS epitope-specific T-cell lines which recognize all six variants used in this study were washed (with PBS, 1% BSA, 0.5 mM EDTA) and then stained with PE-tetramer in the presence of a 100-fold excess of nonbiotinylated pMHC and incubated at 310 K for 30 min. Cells were washed and stained with fluorescein isothiocyanate (FITC) labelled anti-CD8 monoclonal antibody at 277 K for 20 min, washed again, fixed and analyzed by flow cytrometry.
2.3. Protein crystallization
Initial screens to establish crystallization conditions used nanolitre-scale sitting drops (100 nl protein solution plus 100 nl reservoir solution) dispensed by a Cartesian Technologies Microsys MIC4000 (Genomic Solutions; Walter et al., 2003 ▶, 2005 ▶). These experiments used CrystalQuick square-well plates (Greiner) and 90 µl reservoir solution. A total of 672 conditions were tested from a range of commercial kits: Hampton Crystal Screens 1 and 2, Emerald Wizard 1 and 2 (DeCode Genetics), Hampton PEG/Ion Screen, Hampton Grid Screen PEG 6000, Hampton Grid Screen Ammonium Sulfate, Hampton Natrix, Hampton Crystal Screen Cryo, Hampton Grid Screen PEG/LiCl, Hampton Grid Screen NaCl, Hampton Grid Screen MPD, Hampton Quik Screen (sodium/potassium phosphate) and Hampton Index Screen (Hampton Research). Promising conditions were repeated by hand in larger scale (1 µl protein solution plus 1 µl reservoir solution) sitting-drop screens [using microbridges (Harlos, 1992 ▶) in VDX plates (Hampton) containing 500 µl of reservoir], varying the concentrations of salt, buffer, precipitant and pH. After a condition capable of yielding well ordered crystals had been found for one pMHC complexes (pMHC variant GTS1.1; 0.63 M NaH2PO4, 1.17 M K2HPO4 pH 8.2), optimal crystallization conditions for the other variants were established using standard sitting-drop screens (Harlos, 1992 ▶) to fine-tune the salt concentration and pH. Crystals of all the HLA-A11–peptide complexes grew at room temperature in 1.17 M K2HPO4 and 0.63 M NaH2PO4 pH 8.0–8.3.
2.4. Cryoprotection, crystal dehydration and data collection
Cryo methods had to be developed independently for each complex. The cryoprotectants screened included perfluoropolyether oil, sodium malonate and a range of concentrations of glycerol. For cryoprotectant screening, crystals were soaked briefly (1–5 min) in 2 µl cryoprotectant solution, flash-cooled in a cold gas stream and maintained at 100 K. The resultant diffraction quality was assessed using an in-house microfocus X-ray generator (Rigaku, Micromax-007) with either MAR 345 or MAR dtb imaging-plate detectors.
Only one of the pMHC complexes, GTS1.1, yielded diffraction of sufficient quality for data collection from flash-cooled cryoprotected crystals without further manipulation. A second, GTS2.4, was cryoprotected by transfer into a solution of the crystal mother liquor plus 30% glycerol, flash-cooled directly in the cryostream at station BM 10.1 of the Synchrotron Radiation Source (SRS, Daresbury, UK) and, after initial diffraction characterization, annealed by blocking off the cryostream for 2 s. The remaining four complexes, GTS2.1, GTS2.2, GTS2.3 and GTS3.1, had to be mounted on a free-mounting system (FMS, Rigaku, The Woodlands, USA; Kiefersauer et al., 2000 ▶) to allow fine control of humidity prior to flash-cooling. This device allows the crystal to be maintained at a chosen relative humidity at room temperature in a standard cryo-loop. A simple graphical user interface then allows the relative humidity to be manipulated in situ. The ‘start’ humidity for each crystal was defined as the humidity at which a drop of the corresponding mother liquor was maintained at constant size. Following dehydration analysis, crystals were mounted on the FMS either at the start humidity or at a defined humidity below 94.5%, allowed to equilibrate and then covered in 2.9 M sodium malonate cryosolution before being frozen in a cold gas stream and checked for diffraction. Crystals with improved diffraction were stored in liquid nitrogen for subsequent synchrotron data collection. For most crystals the best diffraction was obtained at 93.5% relative humidity, which was slightly below the usual start humidity (Table 2 ▶).
Table 2. Crystallographic data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Peptide | GTS2.1 | GTS2.2 | GTS2.3 | GTS2.4 | GTS1.1 | GTS3.1 |
|---|---|---|---|---|---|---|
| Crystallization pH | 8.0 | 8.3 | 8.1 | 8.3 | 8.2 | 8.2 |
| Cryoprotectant | 2.9 M sodium malonate pH 7.0 | 2.9 M sodium malonate pH 7.0 | 2.9 M sodium malonate pH 7.0 | 30% glycerol with mother liquor | 20% glycerol with mother liquor | 2.9 M sodium malonate pH 7.0 |
| Method of improving crystal diffraction | FMS† | FMS† | FMS† | Annealing | — | FMS† |
| Humidity at which crystal was frozen (%) | 94.5 | 93.5 | 93.5 | 93.5 | ||
| FMS† start humidity (%) | 94.5 | 94.5 | 93.5 | — | — | 94.5 |
| Solvent content (%) | 70.4 | 67.9 | 70.2 | 69.9 | 70.1 | 67.9 |
| Resolution range (Å) | 30–2.80 (2.90–2.80) | 20–2.60 (2.66–2.60) | 25–2.40 (2.47–2.40) | 20–3.20 (3.31–3.20) | 30–2.60 (2.69–2.60) | 20–2.80 (2.86–2.80) |
| No. of reflections collected | 213283 | 206211 | 209829 | 117774 | 222499 | 146030 |
| Unique reflections | 50399 (5025) | 58379 (3648) | 74962 (6075) | 32447 (1567) | 62767 (5948) | 46370 (2872) |
| Redundancy (%) | 4.2 | 3.5 | 2.8 | 3.6 | 3.5 | 3.1 |
| Completeness (%) | 100.0 (100.0) | 99.9 (99.9) | 93.9 (92.0) | 94.4 (45.7) | 99.3 (94.3) | 99.1 (98.1) |
| Rmerge‡ (%) | 9.9 (52.7) | 10.7 (51.9) | 7.5 (52.8) | 14.1 (61.8) | 9.7 (52.1) | 10.5 (47.9) |
| 〈I/σ(I)〉 | 13.9 (2.6) | 10.5 (2.4) | 16.8 (1.7) | 10.9 (1.8) | 12.6 (1.8) | 10.4 (2.3) |
| Space group | C2 | C2 | C2 | C2 | C2 | C2 |
| Unit-cell parameters (Å) | ||||||
| a (Å) | 158.5 | 151.8 | 157.7 | 145.4 | 157.1 | 152.2 |
| b (Å) | 145.7 | 143.6 | 146.3 | 143.6 | 147.1 | 143.9 |
| c (Å) | 105.0 | 102.6 | 104.6 | 113.5 | 104.1 | 101.7 |
| β (°) | 120.2 | 119.8 | 120.2 | 122.3 | 120.1 | 119.7 |
| Unit-cell volume (106 Å3) | 2.096 | 1.941 | 2.086 | 2.003 | 2.081 | 1.935 |
| Source | ESRF ID14-EH2 | SRS BM14.1 | SRS BM14.1 | SRS BM10.1 | ESRF ID23-EH1 | SRS BM14.1 |
| Wavelength (Å) | 0.933 | 1.488 | 1.488 | 1.381 | 0.979 | 1.488 |
| Detector | ADSC Q4 | ADSC Q4 | ADSC Q4 | MAR 225 CCD | MAR 225 CCD | ADSC Q4 |
FMS, free-mounting system (Kiefersauer et al., 2000 ▶).
R
merge = 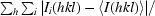
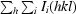 , where Ii(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of reflection hkl.
, where Ii(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of reflection hkl.
Diffraction data for the six complexes were recorded at the beamlines of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and the UK Synchrotron Radiation Source (SRS, Daresbury, UK), as detailed in Table 2 ▶. Diffraction data sets were autoindexed and integrated with DENZO and scaled using SCALEPACK (Otwinowski & Minor, 1997 ▶; http://www.hkl-xray.com) (Table 2 ▶).
3. Results and discussion
3.1. Protein production and functional assays
The yields of refolded complex after size-exclusion chromatography purification were 3–5 times higher for nonbiotinylated pMHCs compared with biotinylated pMHCs, indicating typical refolding efficiencies of 12 and 3%, respectively (data not shown), the former being in line with previously reported values (10–20% depending on peptide and MHC heavy chain) for pMHCs refolded for structural studies (Reid et al., 1996 ▶). Gel electrophoresis indicated that the samples were over 98% pure (Fig. 1 ▶).
Figure 1.

Production of pMHC. pMHCs purified by anion-exchange chromatography were run on 15% SDS–PAGE under reducing conditions. Standard molecular-weight markers (kDa) were run in lane 1, while inclusion bodies of MHC heavy chain and β2m were in lane 2 and 3. Lanes 4–7 show purified GTS1.1, GTS2.2, GTS2.1 and GTS3.1, respectively.
The conformation of the refolded pMHCs was verified by DEIA using monoclonal antibody W6/32 that recognizes a conformational epitope. Furthermore, the T-cell receptor (TCR) binding ability of the refolded pMHCs was tested by a competitive binding assay as shown in Fig. 2 ▶. The refolded pMHCs inhibited PE-tetramers binding to GTS-specific T-cell lines, resulting in reduction of the fluorescent intensity. These demonstrated that the refolded pMHCs retained the functionality of the physiological membrane-bound pMHCs since they had the ability to interact with GTS epitope-specific T-cell lines.
Figure 2.

Tetramer competition assay. The GTS-specific T-cell lines were stained with 1 µg PE-conjugated tetrameric pMHC in the presence (blue) or absence (red) of a 100-fold excess of the refolded pMHC and analyzed by FACS. The histograms show the fluorescent intensity of cells stained with the PE-conjugated tetrameric pMHC complexes.
3.2. Crystallization
672 crystallization conditions were screened in a series of experiments using several batches of the GTS1.1 and GTS3.1 variant pMHCs. Crystallization occurred at room temperature in two major groups of screen conditions; namely, those containing ammonium sulfate or phosphate salts (Table 3 ▶). Initial efforts to collect suitable X-ray data focused on optimized crystals from the ammonium sulfate condition (which was the first of the two crystallization precipitants to be identified). These crystals typically diffracted to 7 Å resolution using both in-house and synchrotron sources. Extensive efforts to improve the diffraction quality (varying the cryoprotection and use of dehydration protocols) provided no consistent route forward. Attention was therefore shifted to the second group of crystallization precipitants to be identified: the phosphate salts.
Table 3. Crystallization conditions that produced pMHC crystals.
| Conditions | |||
|---|---|---|---|
| Salt | Buffer | Additive | Crystallization time |
| 2 M (NH4)2SO4 | 100 mM citrate pH 5.5 | None | 2 d |
| 2 M (NH4)2SO4/200 mM NaCl | 100 mM cacodylate pH 6.5 | None | 2 d |
| 2 M (NH4)2SO4/200 mM Li2SO4 | 100 mM Tris pH 7.0 | None | 2 d |
| 2.4 M (NH4)2SO4 | 1 M MES pH 6.0 | None | 1 d |
| 1.26 M trisodium citrate | 0.09 M Na HEPES pH 7.5 | 10% glycerol | 13 d |
| 1.17 M K2HPO4/0.63 M NaH2PO4 | None | None | 1 d |
| 1.53 M K2HPO4/0.27 M NaH2PO4 | None | None | 1 h |
| 1.73 M K2HPO4/0.072 M NaH2PO4 | None | None | 1 d |
| None | 1.8 M triammonium citrate pH 7.0 | None | 2 d |
A reservoir solution consisting of 0.63 M NaH2PO4 and 1.17 M K2HPO4 pH 8.2 was judged on the basis of visual inspection to be the most promising of the phosphate conditions identified in the nanolitre-scale screens. This condition was therefore used as the basis for crystal optimization in standard microlitre-scale sitting drops. The GTS1.1 variant crystallized within 16 h using this condition and showed Bragg diffraction to better than 3.5 Å resolution when tested on the in-house rotating-anode X-ray source (after cryoprotection by transfer to mother liquor containing 20% glycerol).
Having established a potentially suitable condition for the growth of crystals of suitable quality for structure determination, microlitre-scale crystallization optimizations were carried out for all six pMHC variants. For each pMHC a series of conditions were tested: these conditions were centred on 0.63 M NaH2PO4, 1.17 M K2HPO4 pH 8.2, but varied in salt concentration and pH. Identical salt concentrations but slight differences in pH proved to be essential for the crystallization of the full set of pMHC variants (Table 2 ▶ and Fig. 3 ▶).
Figure 3.

GTS pMHC crystals. Purified pMHCs were crystallized in 0.63 M NaH2PO4, 1.17 M K2HPO4 buffer using sitting-drop vapour diffusion. The buffer pH was varied for each variant as described in Table 2 ▶. All crystals grew at room temperature within 24 h.
Crystallization conditions for the MHC class I molecule HLA-A11 have been reported previously by two groups (Li et al., 2002 ▶; Li & Bouvier, 2004 ▶; Blicher et al., 2005 ▶, 2006 ▶). Bouvier and coworkers reported data for HLA-A11 in complex with two unrelated peptide epitopes from HIV-1 proteins (AIFQSSMTK from reverse transcriptase and QVPLRPMTYK from Nef; Li & Bouvier, 2004 ▶), while Blicher and coworkers (Blicher et al., 2005 ▶, 2006 ▶) detail crystals of two further complexes, those of HLA-A11 with a peptide (KTFPPTEPK) derived from the SARS nucleocapsid and with AIMPARFYPK, a sequence homologue of an epitope in hepatitis B virus DNA polymerase. The crystallization conditions in all these examples were PEG-based and were closely related (the crystallization precipitant was either PEG MME 5000 or PEG 4000 at a concentration of 20–30%). Indeed, PEG-based conditions are typical for the crystallization of pMHC complexes (Garboczi et al., 1994 ▶; Reid et al., 1996 ▶). The nonstandard conditions required for the crystallization of our HLA-A11–peptide complexes indicate that a broad crystallization screen should be considered if initial attempts centred on PEG-based conditions fail to yield pMHC crystals. In particular, the 1.8 M phosphate conditions that yielded useful crystals came from the Hampton Quik Screen (block 5 in our standard set of 480 conditions and usually one of the less successful screens; Walter et al., 2003 ▶, 2005 ▶).
3.3. Crystal lattice-order optimization and X-ray data collection
It was difficult to establish cryo methods for the collection of well ordered Bragg diffraction. Optimal diffraction data for each complex were obtained using different protocols (Table 2 ▶). Two cryoprotectants proved useful, glycerol and sodium malonate, but X-ray data to a suitable resolution for structure determination could only be collected directly from cryoprotected crystals for the GTS1.1 variant pMHC (2.6 Å; Table 2 ▶). The crystals of all five other complexes required annealing/fine control of humidity to optimize the diffraction quality. The results of a simple annealing experiment carried out at station BM 10.1 of the SRS (Daresbury, UK) by blocking off the cryostream for 2 s first alerted us to the significant potential for improvement in diffraction quality (Fig. 4 ▶). In this experiment, the resolution limit for Bragg diffraction from a crystal of the GTS2.4 variant complex was improved dramatically from ∼7 to 3.2 Å. Subsequently, the diffraction quality for crystals of the four remaining complexes, GTS2.1, GTS2.2, GTS2.3 and GTS3.1, was systematically characterized at defined humidity levels using a combination of FMS (Kiefersauer et al., 2000 ▶) and in-house X-ray source/detector. The FMS is still a relatively new device, the principle of which is explained in §2.4. Essentially, by maintaining the crystal in an air stream of defined humidity, the crystal can be dehydrated or rehydrated over time. The FMS goniometer head can be mounted onto standard X-ray equipment so that the diffraction at each humidity level can be recorded.
Figure 4.

Improvement of X-ray diffraction after annealing experiment. The cooling flow from the cryostream to the GTS2.4 crystal was blocked off for 2 s between the measurement of the X-ray diffraction patterns shown in (a) and (b). The images are from a MAR CCD detector on beamline 10.1 of the SRS (Daresbury), with the crystal-to-detector distance set such that the resolution at the edge of the detector was 3.5 Å. The experimental conditions (exposure time, oscillation range etc.) are identical between images, with the exception that the backstop position has altered (the backstop was manually moved out of position to allow access to the crystal for annealing and then repositioned). For subsequent data collection, the detector position was altered to measure data to a somewhat higher resolution limit (3.2 Å, Table 2 ▶).
In this case, we performed full dehydration analyses on only a few crystals and then used fresh crystals to repeat the dehydration steps which resulted in the best diffraction. Crystals were then frozen directly in a standard cold nitrogen-gas stream without testing their diffraction at room temperature and were only exposed to X-rays once they had been frozen (suitable crystals were then stored in liquid nitrogen for subsequent synchrotron data collection). This approach avoids any of the radiation damage which the crystals experience when they are exposed to X-rays at room temperature. Typically, out of ten crystals tested, four showed improved diffraction. This rather low success rate may partly be a consequence of the difficulty in reproducing the freezing protocol exactly (for example, in covering the crystal with the freezing solution and transferring it to the cold gas stream always within the same time interval). It is clear that even with the FMS there is scope for improving the crystal-handling procedures in order to achieve greater reproducibility for sensitive crystals such as these. From Table 2 ▶, it can be seen that for two of the complexes (GTS2.1 and GTS2.3) the optimal humidity level is the start humidity (i.e. that of the crystallization mother liquor); thus, the use of the FMS appears to have allowed us to circumvent residual problems with cryoprotection. For the other two complexes (GTS2.2 and GTS3.1), the optimal humidity is 1% below the start humidity, implying that a slight dehydration of the crystal promotes a beneficial lattice rearrangement.
As expected given their closely related crystallization conditions, the crystals of all six pMHC complexes had essentially identical lattices. The crystals belong to space group C2 and contain three pMHC complexes in the crystallographic asymmetric unit plus 68–70% solvent (the Matthews coefficients are 3.83–4.16 Å3 Da−1; further details including unit-cell parameters are given in Table 2 ▶). It is noteworthy that the percentage of solvent in the crystals does indeed correlate with the protocols applied before data collection. The three crystals subjected to annealing or controlled dehydration all show lower solvent content than those simply cryoprotected or flash-frozen at start humidity on the FMS. In all cases the solvent content is relatively high for a protein crystal, particularly when compared with the values of 48–58% for other peptide–HLA-A11 complexes (Li & Bouvier, 2004 ▶; Blicher et al., 2005 ▶, 2006 ▶) and pMHC complex crystals in general (Garboczi et al., 1994 ▶; Reid et al., 1996 ▶). The diffraction data reported here have resulted in successful structure determinations (phased by molecular replacement) for all six variant complexes. A full analysis of the crystal structures and their implications for the immune response to dengue virus is ongoing and will be reported elsewhere.
4. Conclusions
In our experience, as in many other laboratories, dehydration of protein crystals can provide essential improvements in diffraction quality (reviewed by Heras & Martin, 2005 ▶). For example, a reduction of crystal solvent content from 56 to 48% and a consequent increase in diffraction limit from 3.7 to 2.2 Å allowed the structure of HIV-1 reverse transcriptase to be refined at sufficient resolution to facilitate antiviral drug design (Esnouf et al., 1998 ▶). Similarly significant improvements in resolution (from ∼3.5 to 2.0 Å) were achieved at rather higher solvent content (63% reduced to 56%) for crystals of human semaphorin 4D (Esnouf et al., 2006 ▶). In both these previous examples, the dehydration protocol was based on stepwise increases in the reservoir concentration of a PEG-based precipitant prior to crystal mounting. The development of humidity-controlling systems such as the FMS now provide an alternative route by which to manipulate crystal hydration levels (see, for example, Koch et al., 2004 ▶). The very high level of solvent content in the dengue peptide–HLA-A11 complexes may well have contributed to the difficulties we encountered in optimizing crystal-preparation protocols for cryo X-ray data collection; however, for these examples a systematic investigation of the effects of humidity, facilitated by an FMS, was successful in bringing problematic crystal lattices into sufficient order to allow useful data collection.
Acknowledgments
We are grateful to T. S. Walter for help with the crystallization setups and to the staff of the ESRF and EMBL in Grenoble, France as well as the SRS in Daresbury, UK for assistance with X-ray data collection. We acknowledge use of the crystallization facilities provided by the MRC-funded Oxford Protein Production Facility (OPPF) and The European Commission Integrated Programme (SPINE, grant code QLG2-CT-2002-00988). EYJ is a Cancer Research UK Principal Research Fellow. PM is supported by Senior Research Scholar Program of the Thailand Research Fund (TRF). PC is supported by The Medical Scholar Program, Mahidol University and The Split Mode PhD program (BIOTEC, Thailand). This work was funded by the UK Medical Research Council and The Wellcome Trust.
References
- Altman, J. D., Moss, P. A., Goulder, P. J., Barouch, D. H., McHeyzer-Williams, M. G., Bell, J. I., McMichael, A. J. & Davis, M. M. (1996). Science, 274, 94–96. [Google Scholar]
- Blicher, T., Kastrup, J. S., Buus, S. & Gajhede, M. (2005). Acta Cryst. D61, 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blicher, T., Kastrup, J. S., Pedersen, L. O., Buus, S. & Gajhede, M. (2006). Acta Cryst. F62, 1179–1184. [DOI] [PMC free article] [PubMed]
- Esnouf, R. M., Love, C. A., Harlos, K., Stuart, D. I. & Jones, E. Y. (2006). Acta Cryst. D62, 108–115. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M., Ren, J., Garman, E. F., Somers, D. O’N., Ross, C. K., Jones, E. Y., Stammers, D. K. & Stuart, D. I. (1998). Acta Cryst. D54, 938–953. [DOI] [PubMed] [Google Scholar]
- Garboczi, D. N., Madden, D. R. & Wiley, D. C. (1994). J. Mol. Biol.239, 581–587. [DOI] [PubMed] [Google Scholar]
- Gubler, D. J. (1998). Clin. Microbiol. Rev.11, 480–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, M. G., Kouri, G., Valdes, L., Bravo, J., Alvarez, M., Vazques, S., Delgado, I. & Halstead, S. B. (2000). Am. J. Epidemiol.152, 793–799. [DOI] [PubMed] [Google Scholar]
- Harlos, K. (1992). J. Appl. Cryst.25, 536–538. [Google Scholar]
- Heras, B. & Martin, J. L. (2005). Acta Cryst. D61, 1173–1180. [DOI] [PubMed] [Google Scholar]
- Kiefersauer, R., Than, M. E., Dobbek, H., Gremer, L., Melero, M., Strobl, S., Dias, J. M., Soulimane, T. & Huber, R. (2000). J. Appl. Cryst.33, 1223–1230. [Google Scholar]
- Koch, M., Breithaupt, C., Kiefersauer, R., Freigang, J., Huber, R. & Messerschmidt, A. (2004). EMBO J.23, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. & Bouvier, M. (2004). J. Immunol.172, 6175–6184. [DOI] [PubMed] [Google Scholar]
- Li, L., Promadej, N., McNicholl, J. M. & Bouvier, M. (2002). Acta Cryst. D58, 1195–1197. [DOI] [PubMed] [Google Scholar]
- Mongkolsapaya, J., Dejnirattisai, W., Xu, X. N., Vasanawathana, S., Tangthawornchaikul, N., Chairunsri, A., Sawasdivorn, S., Duangchinda, T., Dong, T., Rowland-Jones, S., Yenchitsomanus, P. T., McMichael, A., Malasit, P. & Screaton, G. (2003). Nature Med.9, 921–927. [DOI] [PubMed] [Google Scholar]
- Mongkolsapaya, J., Duangchinda, T., Dejnirattisai, W., Vasanawathana, S., Aviruthnan, P., Jairungsri, A., Khemnu, N., Tangthawornchaikul, N., Chotiyarnwong, P., Sae-Jang, K., Koch, M., Jones, Y., McMichael, A., Xu, X., Malasit, P. & Screaton, G. (2006). J. Immunol.176. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reid, S. W., Smith, K. J., Jakobsen, B. K., O’Callaghan, C. A., Reyburn, H., Harlos, K., Stuart, D. I., McMichael, A. J., Bell, J. I. & Jones, E. Y. (1996). FEBS Lett.383, 119–123. [DOI] [PubMed] [Google Scholar]
- Shields, M. J. & Ribaudo, R. K. (1998). Tissue Antigens, 51, 567–570. [DOI] [PubMed] [Google Scholar]
- Walter, T. S., Diprose, J., Brown, J., Pickford, M., Owens, R. J., Stuart, D. I. & Harlos, K. (2003). J. Appl. Cryst.36, 308–314. [Google Scholar]
- Walter, T. S., Diprose, J. M., Mayo, C. J., Siebold, C., Pickford, M. G., Carter, L., Sutton, G. C., Berrow, N. S., Brown, J., Berry, I. M., Stewart-Jones, G. B. E., Grimes, J. M., Stammers, D. K., Esnouf, R. M., Jones, E. Y., Owens, R. J., Stuart, D. I. & Harlos, K. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2002). Dengue and Dengue Haemorrhagic Fever. World Health Organization Fact Sheet 117. http://www.who.int/mediacentre/factsheets/fs117/en/.