

A RecB-like nuclease from the archaeon Pyrococcus abyssi was expressed, purified and crystallized. The crystals belong to the orthorhombic space group C2221 with a = 81.5, b = 159.8, c = 100.8 Å, and a native data set was collected to 2.65 Å resolution.
Keywords: nucleases, RecB, Pyrococcus abyssi
Abstract
Nucleases are required to process and repair DNA damage in living cells. One of the best studied nucleases is the RecB protein, which functions in Escherichia coli as a component of the RecBCD enzyme complex that amends double-strand breaks in DNA. Although archaea do not contain the RecBCD complex, a RecB-like nuclease from Pyrococcus abyssi has been cloned, expressed and purified. The protein was crystallized by the sitting-drop vapour-diffusion method using polyethylene glycol 8000 as the precipitant. The crystals belong to the orthorhombic space group C2221, with unit-cell parameters a = 81.5, b = 159.8, c = 100.8 Å. Self-rotation function and native Patterson map calculations revealed that there is a dimer in the asymmetric unit with its local twofold axis running parallel to the crystallographic twofold screw axis. The crystals diffracted to about 2 Å and a complete native data set was collected to 2.65 Å resolution.
1. Introduction
As all cells have to maintain their genomes intact, all organisms and particularly those that thrive under extreme conditions contain several nucleases that are required for the processing and reparation of DNA damage. Recently, several new nucleases have been identified in bacteria and eukarya (Aravind et al., 2000 ▶). These nucleases function either in association with DNA polymerase or autonomously and are often involved in improving the fidelity of the replisome. One of the best studied nucleases is the RecB protein, which is associated with the RecBCD complex in Escherichia coli. This complex participates in the repair of double-strand breaks by homologous recombination. Recent structural studies (Singleton et al., 2004 ▶) have indicated that the RecC subunit of this complex creates a fork structure that separates the DNA strands and directs them towards the active site of the RecB nuclease (exonuclease V family; Shevelev & Hubscher, 2002 ▶).
Although archaea do not seem to contain the RecBCD complex, many archaeal genomes encode proteins that have been annotated as ‘predicted nuclease of the RecB family’ (Aravind et al., 2000 ▶; Kinch et al., 2005 ▶). In the genome of Pyrococcus abyssi, a hyperthermophilic euryarchaeon, one of these putative nucleases is encoded by the open reading frame PAB2263. Preliminary studies indicate that PAB2263 is an exonuclease (data not shown). It is also of note that PAB2263 and its orthologues are a part of a superfamily that, in addition to RecB nucleases, includes several restriction endonucleases, archaeal Holliday junction resolvase and XPF/Rad1/Mus81 nuclease. Many of these proteins contain (GV)hhD and (DE)hK signatures and a conserved tyrosine near the C-terminus (Wang et al., 2000 ▶) that presumably participate in DNA degradation. However, the lack of readily detectable sequence similarity outside of their active sites has complicated their characterization and functional annotation.
To provide a structural description of these nuclease families, we have overexpressed, purified and crystallized PAB2263 from P. abyssi and collected preliminary diffraction data from these crystals. This study is motivated by the belief that structural analysis of this putative nuclease will help in understanding the functional role of this protein in cellular physiology.
2. Materials and methods
2.1. Cloning and expression
The gene encoding P. abyssi PAB2263 was amplified using 30 cycles of PCR using chromosomal DNA as template. A cis-expressed pQE-80L (Qiagen) expression vector containing isopropyl β-d-thiogalactopyranoside (IPTG) inducible bacteriophage T5 promoter was used to express all proteins. PAB2263 was cloned using BamHI and PstI restriction sites carried by the oligonucleotides used for amplification (all primer sequences used during this work are available upon request). The resulting expression construct encodes the full-length protein carrying a six-histidine tag at its N-terminus. The His tag is directly attached to the N-terminus of the protein without a tether.
The expression construct was transformed into E. coli BL21-CodonPlus-RIL strain (Stratagene) and cultured on LB medium plates containing 100 µg ml−1 ampicillin. Protein expression was induced by adding IPTG to the cell cultures to a final concentration of 0.5 mM when cell growth reached the exponential phase. After 2 h induction, the cells were collected by centrifugation and processed as indicated below.
2.2. Cell lysis and protein purification
Cell lysis was performed in buffer A (30 mM HEPES pH 8, 300 mM NaCl) by several freeze–thaw cycles followed by brief sonication to decrease the viscosity of the supernatant. Cellular debris was eliminated by centrifugation. All chromatographic procedures were performed with an ÄKTA FPLC system at 283 K (Amersham Biosciences). Tagged proteins were purified on immobilized Ni–NTA agarose (Amersham), followed by purification on an S-200 gel-filtration column (Amersham). All protein samples were analyzed for purity and integrity using 11% SDS–PAGE and by MALDI–TOF analysis (Innova Proteomics, France). The purified protein was concentrated using a Microsep 10K centrifugal device (Pall Life Sciences). It was found that a relatively high salt concentration (0.5–0.6 M NaCl) was necessary to keep the protein fully soluble.
2.3. Crystallization
Crystallization conditions were searched for by the sitting-drop vapour-diffusion method at 293 K using Hampton Research screening kits and Cryschem plates (Hampton Research). Small crystals were observed with 0.1 M HEPES pH 7.5 or 0.1 M Tris–HCl pH 8.5 as the buffer when various polyethylene glycols were present as precipitants, including PEG MME (polyethylene glycol monomethyl ether) 2000, PEG 3350, PEG 6000, PEG 8000 and PEG 20 000. Almost all of the crystals appeared from precipitates, with an initial protein concentration in the range 1–6.6 mg ml−1. The crystallization conditions were optimized by a fine grid search with combinations of buffers and polyethylene glycols at various concentrations. Crystals suitable for X-ray analysis were obtained by mixing 2 µl protein solution (at a concentration of 4 mg ml−1 in 30 mM HEPES pH 8.0 and 0.57 M NaCl) with 2 µl reservoir solution consisting of 0.1 M HEPES pH 7.5 and 6% PEG 8000 to make the initial droplet, which was equilibrated against 0.5 ml reservoir solution. Prior to data collection at cryotemperatures, the crystals were briefly immersed in a cryoprotectant solution containing 6% PEG 8000, 15% glycerol and 0.1 M HEPES pH 7.5.
2.4. Data collection and preliminary X-ray analysis
A native data set was collected at a cryotemperature of around 100 K on beamline BW7A at the EMBL Outstation, DESY (Hamburg, Germany) using a MAR CCD detector. The wavelength was 0.9050 Å. The data-collection strategy was determined using the program BEST (Popov & Bourenkov, 2003 ▶). The data set consisted of 344 images collected over four different oscillation ranges with a step of 0.31° or 0.65° at a distance of 210 mm. The data were processed using the HKL package (Otwinowski & Minor, 1997 ▶).
A self-rotation function was calculated to check the local twofold symmetry of the molecules in the asymmetric unit using data in the resolution range 15–4 Å. The Patterson vectors used in the analysis have a length of between 4 and 45 Å, with the origin peak removed. To further localize and determine the orientation of the noncrystallographic twofold symmetry, a native Patterson function was calculated using data between 20 and 4 Å resolution, followed by analysis of the vector peaks on Harker sections. The calculations were carried out with the CNS program suite (Brünger et al., 1998 ▶).
3. Results and discussion
Single crystals appeared in the crystallization droplets within one week. They grew to a maximum dimension of about 0.3 mm in two weeks (Fig. 1 ▶). The crystals diffracted anisotropically to a maximum resolution of about 2 Å in the strongest direction using synchrotron radiation. As a result, an initial native data set was collected to an effective resolution of 2.65 Å. Autoindexing of the diffraction data suggested the existence of a C-centred orthorhombic lattice. Inspection of the systematic extinctions and the Laue symmetry of the diffraction pattern revealed that the crystal belonged to space group C2221. The unit-cell parameters of the crystal are a = 81.5, b = 159.8, c = 100.8 Å. Data-collection and processing statistics are shown in Table 1 ▶. Weakened reflections at high resolutions and the anisotropic nature of the crystal led to an R merge value that was a little higher for the data in the outermost resolution shell.
Figure 1.

P. abyssi nuclease crystal. The maximum dimension of this crystal is about 0.3 mm.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Beamline | BW7A, DESY Hamburg |
| Wavelength (Å) | 0.9050 |
| Space group | C2221 |
| Unit-cell parameters (Å, °) | a = 81.5, b = 159.8, c = 100.8, α = β = γ = 90 |
| Resolution (Å) | 15–2.65 (2.68–2.65) |
| No. of observations | 111703 |
| Unique reflections | 35562 |
| Average redundancy | 3.1 (3.2) |
| Completeness (%) | 96.8 (99.7) |
| Average I/σ(I) | 17.8 (2.6) |
| Rmerge† (%) | 5.3 (61.6) |
R
merge = 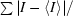
 .
.
With a six-residue His tag attached at the N-terminus, the protein has a calculated molecular weight of 29 654 Da. Assuming the presence of one dimer in the asymmetric unit, the crystal has a solvent content of 55.5%, corresponding to a V M value of 2.76 Å3 Da−1, which is within the commonly observed range of values found for proteins (Matthews, 1968 ▶). The self-rotation function calculation, however, shows no additional twofold symmetry peaks besides the crystallographic symmetry (Fig. 2 ▶). As the local axis could run parallel to a crystallographic twofold axis and thus would not be separately visible in a self-rotation map, a native Patterson function was calculated, which reveals a large non-origin peak resulting from the translational vectors relating the corresponding atoms in the subunits of the dimer (Fig. 3 ▶). This peak has a height of 36% of the Patterson origin peak and is located at (u, v, w) = (0, 0.4750, 0.5000). It is thus concluded that the twofold noncrystallographic axis is parallel to the crystallographic twofold screw axis in the c direction at an approximate position (x, y) = (0, 0.2375). Preliminary gel-filtration experiments suggested that the protein exists as a tetramer in solution. It is therefore tempting to speculate that the tetramers are probably built up in the crystal by relating neighbouring dimers by crystallographic twofold symmetry operations. Expression of the selenomethionine-substituted protein and structure determination by the multiple-wavelength anomalous diffraction method (MAD) are under way.
Figure 2.

Plot of the self-rotation function at κ = 180°. The self-rotation function was calculated using data between 15 and 4 Å. Only peaks corresponding to the crystallographic twofold symmetry are visible, which suggests that the noncrystallographic axis may be orientated parallel to the crystallographic axis. The map is contoured from the 1.8σ level in steps of 0.9σ.
Figure 3.

Harker section (u, v, 1/2w) of the native Patterson map calculated using data between 20 and 4 Å. The large peak located at (u, v) = (0, 0.4750) corresponds to the translation vector between corresponding atoms in the subunits of the dimer in the asymmetric unit, whose axis is parallel to the crystallographic twofold screw axis. The map is contoured from 1.0σ in steps of 1.0σ.
Acknowledgments
We thank the EMBL Outstation at DESY (Hamburg, Germany) for providing access to synchrotron beamline BW7A and Dr Alexander Popov for technical help during data collection. This work was supported by the European Commission project REPBIOTECH.
References
- Aravind, L., Makarova, K. S. & Koonin, E. V. (2000). Nucleic Acids Res.28, 3417–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, N., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Kinch, L. N., Ginalski, K., Rychlewski, L. & Grishin, N. V. (2005). Nucleic Acids Res.33, 3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Popov, A. N. & Bourenkov, G. P. (2003). Acta Cryst. D59, 1145–1153. [DOI] [PubMed] [Google Scholar]
- Shevelev, I. V. & Hubscher, U. (2002). Nature Rev. Mol. Cell Biol.3, 364–376. [DOI] [PubMed]
- Singleton, M. R., Dillingham, M. S., Gaudier, M., Kowalczykowski, S. C. & Wigley, D. B. (2004). Nature (London), 432, 187–93. [DOI] [PubMed] [Google Scholar]
- Wang, J., Chen, R. & Julin, D. A. (2000). J. Biol. Chem.275, 507–513. [DOI] [PubMed] [Google Scholar]