

Preliminary crystallographic data are reported for 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) from A. nitroguajacolicus Rü61a.
Keywords: oxygenase, cofactor-free, α/β-hydrolase, 1H-3-hydroxy-4-oxoquinaldine, SAD
Abstract
1H-3-Hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) is a cofactor-devoid dioxygenase that is involved in the anthranilate pathway of quinaldine degradation. HOD has been proposed to belong to the α/β-hydrolase-fold superfamily of enzymes. N-terminally His6-tagged HOD has been crystallized by the hanging-drop vapour-diffusion method using sodium/potassium tartrate as a precipitant and CuCl2 as an additive. The structure was solved by the single anomalous dispersion (SAD) technique using data collected to 3.5 Å resolution at the Cu absorption peak wavelength. The crystals belong to the primitive tetragonal space group P43212, with unit-cell parameters a = b = 153.788, c = 120.872 Å.
1. Introduction
In several aerobic metabolic pathways, O2 is incorporated into organic compounds through monooxygenase- or dioxygenase-catalyzed reactions (Hayaishi, 1974 ▶). For instance, oxygenation is often used to make lipids and particularly aromatic molecules amenable to further biochemical transformations. 1H-3-Hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD; EC 1.13.11.48) from Arthrobacter nitroguajacolicus Rü61a is a 32 kDa enzyme that is involved in the anthranilate pathway of quinaldine degradation (Bauer et al., 1996 ▶). It catalyses the O2-dependent N-heteroaromatic ring cleavage of 1H-3-hydroxy-4-oxoquinaldine to N-acetylanthranilate and carbon monoxide (Fig. 1 ▶).
Figure 1.

2,4-Dioxygenolytic ring cleavage of 1H-3-hydroxy-4-oxoquinaldine catalysed by HOD. Oxygen atoms from dioxygen are shown in larger bold font. This figure was created with MarvinSketch (ChemAxon).
Sequence analysis and secondary-structure prediction indicate that HOD belongs to the large superfamily of α/β-hydrolase-fold enzymes (Fischer et al., 1999 ▶; Nardini & Dijkstra, 1999 ▶). HOD displays no sequence similarities to other known oxygenases, except for the 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase (QDO; EC 1.13.11.47) involved in 1H-4-oxoquinoline degradation by Pseudomonas putida 33/1 (Qi et al., 2007 ▶). QDO catalyses a reaction very similar to that catalysed by HOD; namely, the dioxygenolytic N-heteroaromatic ring cleavage of 1H-3-hydroxy-4-oxoquinoline to N-formylanthranilate and carbon monoxide (Bauer et al., 1996 ▶). HOD and QDO share 37% identity at the sequence level and these two enzymes are the only dioxygenases that have been proposed to be members of the α/β-hydrolase-fold enzymes.
Oxygenases usually depend on a transition metal and/or organic cofactors for activity. This is a consequence of the fact that a direct reaction between the triplet ground-state O2 and singlet ground-state substrates to produce singlet-state products implies a violation of the conservation of the total angular momentum (Hamilton, 1974 ▶). It is therefore a low-probability event. The presence of cofactors overcomes the spin-forbiddenness of the process. The mechanism used by nonhaem iron-dependent (Que, 1999 ▶), copper-dependent (Steiner et al., 2002 ▶), flavin-dependent and pterin-dependent (Massey, 1994 ▶; Palfey et al., 1995 ▶) oxygenases have been studied and various reviews are available that discuss the general mechanisms of oxygen and substrate activation in enzymatic oxygenation reactions. Biochemical and spectroscopic studies have shown that neither HOD nor QDO contain organic cofactors or stoichiometric amounts of any metal (Bauer et al., 1996 ▶; Fetzner, 2002 ▶). The mechanism(s) employed by these dioxygenases are therefore very interesting from the viewpoint of fundamental enzymology.
The availability of three-dimensional information should increase our understanding of the catalytic mechanism employed by these intriguing cofactor-devoid dioxygenases as well as provide direct evidence about their fold. In this account, we report the crystallization and preliminary X-ray analysis of HOD.
2. Materials and methods
2.1. Cloning and expression
The hod gene from genomic DNA of A. nitroguajacolicus Rü61a was isolated and cloned by PCR amplification as described in Frerichs-Deeken et al. (2004 ▶). For protein overexpression in Escherichia coli, the target gene was inserted into the pQE30 vector (Qiagen) at the BamHI and SalI restriction sites. An N-terminal His6 fusion tag of sequence MRGSHHHHHHGS was added to the gene product in order to facilitate protein purification. The recombinant vector containing the target gene was transformed in competent E. coli M15[pREP4] cells by electroporation.
Transformed cells were grown overnight in 10 ml Luria–Bertani (LB) medium supplemented with 50 µg ml−1 kanamycin and 100 µg ml−1 ampicillin at 298 K. 400 µl of the overnight starter culture was then transferred into 400 ml LB medium supplemented with antibiotics and incubated at 310 K until the OD at 600 nm reached 0.5. After decreasing the temperature to 301 K, expression of His6-HOD was induced with 0.5 mM IPTG; the cells were allowed to grow for a further 20 h and were then harvested by centrifugation at 3500g and 298 K for 15 min.
2.2. Purification
Cells were resuspended in 50 mM Tris–HCl pH 8.0, 200 mM NaCl, 20 mM imidazole supplemented with 1× protease-inhibitor cocktail (Calbiochem) lysis buffer and lysed by pulsed sonication on ice. Cell debris was pelleted by centrifugation at 44 000g and 277 K for 45 min. The supernatant was filtered through 0.22 µm filters and loaded onto a 5 ml Ni-loaded His-Trap (GE Healthcare) column previously equilibrated with 50 mM Tris–HCl pH 8.0, 200 mM NaCl, 20 mM imidazole and washed with this buffer until a constant A 280 was observed. Protein was eluted with a gradient to 50 mM Tris–HCl pH 8.0, 200 mM NaCl, 500 mM imidazole and fractions containing HOD were pooled.
After dialysis overnight against 50 mM Tris–HCl pH 8.0, 5 mM KCl, 2 mM EDTA, the His6-HOD solution was loaded onto a Resource-Q (GE Healthcare) ion-exchange column previously equilibrated with dialysis buffer. HOD was eluted with a gradient to 50 mM Tris–HCl pH 8.0, 600 mM KCl, 2 mM EDTA. The protein was concentrated and further equilibrated in 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, 1 mM DTT (storage buffer) for storage at 193 K.
Frerichs-Deeken et al. (2004 ▶) have shown that HOD has a tendency to form a mixture of monomeric and dimeric species and that dimerization can be prevented by mutating Cys69 to Ser. The HOD C69S variant has catalytic properties that are identical to those of wild-type HOD (Frerichs-Deeken et al., 2004 ▶). Therefore, we produced the His6-HOD C69S variant as well as the inactive His6-HOD C69S/H251A double variant using the QuikChange (Stratagene) mutagenesis system. The His6-HOD C69S/H251A double variant is catalytically inactive owing to the His→Ala substitution of the essential residue His251 (Frerichs-Deeken et al., 2004 ▶). Both His6-HOD variants were purified according to the protocol described above. An SDS–PAGE gel of purified His6-HOD C69S/H251A is shown in Fig. 2 ▶. On average, the typical yield of pure protein material is approximately 30 mg per litre of culture.
Figure 2.

SDS–PAGE profile of the A. nitroguajacolicus Rü61a His6-HOD C69S/H251A protein. Lane 1, molecular-weight markers. The molecular weight of each protein band is shown in kDa on the left of the figure. Lane 2, purified His6-HOD C69S/H251A protein.
2.3. Crystallization
Owing to their more homogeneous behaviour in solution, crystallization trials were mainly carried out using the His6-HOD variants described in §2.2. Crystallization conditions were established with the hanging-drop vapour-diffusion technique using 24-well Linbro plates. All experiments were carried out at a constant temperature of 291.15 K using a protein:precipitant ratio of 1.0. Initial crystallization trials of His6-HOD C69S/H251A at concentrations of up to 50 mg ml−1 in storage buffer using various commercial screens (Crystal Screen, Crystal Screen 2, Index, SaltRx, PEG/Ion from Hampton Research) as well as noncommercial versions of the Clear Crystal Strategy 1 and Clear Crystal Strategy 2 screens (Brzozowski & Walton, 2001 ▶) failed to produce crystals. However, analysis of the results obtained from the crystallization trials using the various kits indicated salts as being more likely to produce crystals compared with organic precipitants. Crystallization conditions containing salts generally gave light precipitates, whereas organic precipitants tended to yield amorphous precipitates. Amorphous precipitates were also often observed in crystallization conditions buffered at pH values below 5.0–5.5. Screening of several salt precipitants (ammonium citrate, ammonium tartrate, sodium/potassium tartrate, sodium malate, sodium malonate, sodium formate, ammonium nitrate, ammonium sulfate, sodium acetate, sodium chloride, lithium sulfate) at various concentrations yielded ‘sea-urchin’-like microcrystals using 1.8 M sodium/potassium tartrate as a precipitant and His6-HOD C69S/H251A in storage buffer at a concentration of 50 mg ml−1 (Fig. 3 ▶ a). Identical microcrystals were also obtained under the same conditions using the His6-HOD C69S variant.
Figure 3.

Crystals of His6-HOD C69S/H251A obtained as described in §2.3.
Further crystal optimization was carried out using the His6-HOD C69S/H251A variant only. Single crystals were obtained by optimizing the precipitant and protein concentration as well as the pH used to obtain the microcrystals shown in Fig. 3 ▶(a). The single crystals shown in Fig. 3 ▶(b) (crystal form A) were obtained using His6-HOD C69S/H251A at a concentration of 150 mg ml−1 in storage buffer in the presence of a reservoir composed of 1.65 M sodium/potassium tartrate and 0.1 M HEPES pH 7.0. SeMet-substituted His6-HOD C69S/H251A failed to crystallize under identical conditions. The His6-HOD C69S/H251A crystals obtained as described above diffracted to 3.15 Å resolution at the ID14-EH1 beamline (ESRF, Grenoble). Analysis of cumulative intensity distribution plots (data not shown) from diffraction data (Table 1 ▶, crystal form A) revealed these crystals, which appeared to belong to point group P422, exhibit twinning.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution bin.
| Data set | Crystal form A (Fig. 3 ▶b) | Crystal form B (Fig. 3 ▶d) |
|---|---|---|
| Beamline | ID14-EH1 (ESRF) | BM30 (ESRF) |
| Wavelength (Å) | 0.935 | 1.3783 |
| Detector | ADSC Q210 CCD | MAR CCD (165 mm) |
| Crystal-to-detector distance (mm) | 265 | 180 |
| Exposure time per degree of rotation (s) | 25 | 80 |
| Space group | Processed in P422 | P43212 |
| Unit-cell parameters (Å) | a = b = 165.77, c = 44.53 | a = b = 153.79, c = 120.87 |
| Resolution limits† (Å) | 40.0–3.15 (3.26–3.15) | 30.00–3.50 (3.63–3.50) |
| I/σ(I) | 17.9 (4.8) | 22.0 (4.4) |
| Observations | 80435 | 300495 |
| Independent reflections | 11373 (1102) | 17555 (1417) |
| Overall redundancy | 7.07 (6.7) | 17.1 (9.4) |
| Rsym† (%) | 9.9 (41.8) | 11.6 (37.2) |
| Completeness (%) | 99.9 (100.0) | 95.0 (78.5) |
| Molecules per ASU | 2‡ | 3 |
| Solvent content (%) | 48.5 | 67.0 |
R
sym = 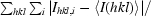
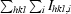 , where 〈Ihkl〉 is the mean intensity of a set of equivalent reflections and I
hkl,i is the ith measurement of the reflection with Miller indices hkl.
, where 〈Ihkl〉 is the mean intensity of a set of equivalent reflections and I
hkl,i is the ith measurement of the reflection with Miller indices hkl.
Given the twinned nature of crystal form A, predictions based on the Matthews coefficient analysis (Matthews, 1968 ▶) give four protein molecules per ASU, assuming the real point group to be either P4 or P222.
In parallel with attempts to solve the structure of His6-HOD C69S/H251A from twinned data using the molecular-replacement technique with various members of the α/β-hydrolase-fold family as templates, we also tried to overcome the twinning problem by screening crystallization additives using the Additive Screening kit (Hampton Research). We found that the addition of 10 mM CuCl2 to the 1.65 M sodium/potassium tartrate, 0.1 M HEPES pH 7.0 crystallization mixture yielded small His6-HOD C69S/H251A crystals of different morphology (Fig. 3 ▶ c). Optimization of the additive concentration led to crystals of square bipyramidal shape (crystal form B; Fig. 3 ▶ d) which typically grow to dimensions of 350 × 350 × 200 µm over three weeks. These His6-HOD C69S/H251A crystals grow from a protein solution at 150 mg ml−1 in storage buffer in the presence of 1.65 M sodium/potassium tartrate, 0.1 M HEPES pH 7.0 and 30 mM CuCl2.
2.4. X-ray diffraction
His6-HOD C69S/H251A crystals grown from 1.65 M sodium/potassium tartrate, 0.1 M HEPES pH 7.0 and 30 mM CuCl2 were transferred for a few seconds into a reservoir containing 1.7 M sodium/potassium tartrate, 0.1 M HEPES pH 7.0, 30 mM CuCl2 and 15%(v/v) glycerol and vitrified in liquid nitrogen for data collection. Although the high salt concentration present in the reservoir allows a high degree of cryoprotection, we observed improved behaviour in the presence of glycerol.
We reasoned that the crystallization of His6-HOD C69S/H251A in the presence of a high concentration of Cu2+ ions might have resulted in heavy atoms bound to the histidine tag and that these might be useful for phasing purposes. A single anomalous dispersion (SAD) data set (Table 1 ▶, crystal form B) was collected at 3.5 Å resolution using synchrotron radiation at the Cu absorption edge (1.3783 Å). Crystallographic data for both HOD crystal forms were processed using the HKL suite (Otwinowski & Minor, 1997 ▶). Analysis of intensity statistics revealed that His6-HOD C69S/H251A crystals grown in the presence of CuCl2 no longer exhibited signs of twinning.
3. Results
Data from HOD crystal form B were used for structure solution. In order to solve the structure of His6-HOD C69S/H251A, the graphical user interface HKL2MAP (Pape & Schneider, 2004 ▶) was used to run several programs (SHELXC, SHELXD, SHELXE) from the SHELX suite. After data preparation with SHELXC (Sheldrick, 2003 ▶), the substructure-solution program SHELXD (Schneider & Sheldrick, 2002 ▶) easily located six high-occupancy Cu atoms in 30 of the 100 phase trials performed. These sites were then directly passed to SHELXE (Sheldrick, 2002 ▶) for phasing and density modification (solvent content set to 65%). A clear difference in contrast was seen between the original and inverted-hand enantiomorphs. Inspection of the electron-density maps revealed regions of correct right-hand helices in space group P43212 which could be fitted to helical segments using the ARP/wARP HelixBuild module (Morris et al., 2004 ▶). Model building using maps calculated from SHARP (Bricogne et al., 2003 ▶) improved phases is nearly complete. We anticipate that the Cu ions used for phasing bind to the N-terminal hexahistidine tags available and that HOD adopts the predicted α/β-hydrolase-fold topology.
Acknowledgments
The staff scientists operating the beamlines ID14-EH1 and BM30 (European Molecular Biology Laboratory, Grenoble) are acknowledged for their help during the experiments. The York Structural Biology Laboratory (University of York, UK) and the Musacchio laboratory (IFOM-IEO Campus, Milan, Italy) are also thanked for their support.
References
- Bauer, I., Max, N., Fetzner, S. & Lingens, F. (1996). Eur. J. Biochem.240, 576–583. [DOI] [PubMed] [Google Scholar]
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. (2003). Acta Cryst. D59, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Brzozowski, A. M. & Walton, J. (2001). J. Appl. Cryst.34, 97–101. [Google Scholar]
- Fetzner, S. (2002). Appl. Microbiol. Biotechnol.60, 243–257. [DOI] [PubMed] [Google Scholar]
- Fischer, F., Künne, S. & Fetzner, S. (1999). J. Bacteriol.181, 5725–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerichs-Deeken, U., Ranguelova, K., Kappl, R., Huttermann, J. & Fetzner, S. (2004). Biochemistry, 43, 14485–14499. [DOI] [PubMed] [Google Scholar]
- Hamilton, G. A. (1974). Molecular Mechanisms of Oxygen Activation, edited by O. Hayaishi, pp. 405–451. New York: Academic Press.
- Hayaishi, O. (1974). Editor. Molecular Mechanisms of Oxygen Activation, pp. 1–28. New York: Academic Press.
- Massey, V. (1994). J. Biol. Chem.269, 22459–22462. [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Morris, R. J., Zwart, P. H., Cohen, S., Fernandez, F. J., Kakaris, M., Kirillova, O., Vonrhein, C., Perrakis, A. & Lamzin, V. S. (2004). J. Synchrotron Rad.11, 56–59. [DOI] [PubMed] [Google Scholar]
- Nardini, M. & Dijkstra, B. W. (1999). Curr. Opin. Struct. Biol.9, 732–737. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Palfey, B. A., Ballou, D. P. & Massey, V. (1995). Active Oxygen in Biochemistry, edited by J. S. Valentine, C. S. Foote, A. Greenberg & J. F. Liebman, pp. 37–83. London: Blackie.
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst.37, 843–844. [Google Scholar]
- Qi, R., Fetzner, S. & Oakley, A. J. (2007). Acta Cryst. F63, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que, L. Jr (1999). Bioinorganic Catalysis, edited by J. Reedijk & E. Bouwman, pp. 269–321. New York: Marcel Dekker.
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M. (2002). Z. Kristallogr.217, 644–650.
- Sheldrick, G. M. (2003). SHELXC. University of Göttingen, Göttingen, Germany.
- Steiner, R. A., Kalk, K. H. & Dijkstra, B. W. (2002). Proc. Natl Acad. Sci. USA, 99, 16625–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]