

A 3.02 Å crystal structure of native GroEL from E. coli is presented.
Keywords: GroEL, chaperone proteins, ATP-binding proteins
Abstract
GroEL is a member of the ATP-dependent chaperonin family that promotes the proper folding of many cytosolic bacterial proteins. The structures of GroEL in a variety of different states have been determined using X-ray crystallography and cryo-electron microscopy. In this study, a 3.02 Å crystal structure of the native GroEL complex from Escherichia coli is presented. The complex was purified and crystallized in the absence of potassium ions, which allowed evaluation of the structural changes that may occur in response to cognate potassium-ion binding by comparison to the previously determined wild-type GroEL structure (PDB code 1xck), in which potassium ions were observed in all 14 subunits. In general, the structure is similar to the previously determined wild-type GroEL crystal structure with some differences in regard to temperature-factor distribution.
1. Introduction
GroEL is a member of the ATP-dependent chaperonin family that, along with its binding partner GroES, promotes the folding of a wide range of proteins in bacteria (Houry et al., 1999 ▶). GroEL has been the subject of numerous structural studies (Braig et al., 1994 ▶, 1995 ▶; Boisvert et al., 1996 ▶; Chen & Sigler, 1999 ▶; Wang & Boisvert, 2003 ▶; Bartolucci et al., 2005 ▶; Ranson et al., 2001 ▶). The GroEL complex consists of 14 identical subunits arranged into two heptameric rings that associate with each other in a back-to-back manner. Each subunit can be divided into three functional domains termed apical, intermediate and equatorial. The apical domain captures unfolded protein substrates and binds GroES, an event that leads to the encapsulation of the substrate protein. The equatorial domain contains the ATP-binding site and forms contacts between the two heptameric rings. Linking the apical and equatorial domains is the intermediate domain, which is flanked by hinge regions that allow movement of the protein in response to ATP and GroES binding (Fenton & Horwich, 1997 ▶). The binding of ATP to the GroEL equatorial domain is a requirement for the binding of GroES (Chandrasekhar et al., 1986 ▶) and the binding of ATP to a particular subunit promotes ATP binding to the other subunits of the same ring and inhibits ATP binding to the subunits of the opposite ring. Coordinated ATP hydrolysis within one ring results in the release of the protein substrate. It has previously been shown that potassium ions are required for ATP hydrolysis by GroEL (Viitanen et al., 1990 ▶) and that potassium ion binding alters the affinity of GroEL for ATP (Todd et al., 1993 ▶). A crystal structure of the GroEL complex with ATP, Mg2+ and K+ bound to all 14 subunits revealed that potassium ions are involved in binding the triphosphate moiety of ATP (Wang & Boisvert, 2003 ▶). It is not clear whether the alteration in ATP affinity in the presence of potassium ions solely arises from electrostatic effects or whether potassium ion binding produces conformational changes that facilitates subsequent ATP binding. In order to evaluate the latter possibility, we crystallized and determined the structure of wild-type apo GroEL in the absence of potassium ions and performed a structural comparison to the previously determined wild-type GroEL structure that contains potassium ions in all 14 cognate binding sites. The native GroEL complex from Escherichia coli, which was isolated using metal-affinity chromatography by virtue of a tight association with a recombinantly expressed ten-histadine-tagged protein, was used in the crystallization experiments presented in this paper.
2. Experimental procedures
2.1. Purification and crystallization
During the course of bacterial protein-expression experiments, we found that GroEL copurified from Ni2+–NTA resin with a maltose-binding protein ten-histidine retinal pigmented epithelium-specific protein (MBP-His10-RPE65) fusion that was expressed in Rosetta 2 (DE3 pLysS) cells (Novagen, San Diego, CA, USA). RPE65 (gi:55775677) was cloned into a modified pMAL expression plasmid (New England Biolabs, Ipswich, MA, USA) containing a tobacco etch virus (TEV) protease site and a His10 tag between the regions encoding MBP and RPE65 (Kristelly et al., 2003 ▶). 6 l of LB media supplemented with 0.3% glucose were each inoculated with 10 ml of an overnight culture and grown at 310 K until an OD600nm of 0.4–0.6 was achieved, at which point the temperature was lowered to 298 K and protein expression was induced with 100 µM IPTG. 4 h after induction, E. coli cells were harvested by centrifugation, flash-cooled in liquid nitrogen and stored at 193 K until use. The bacterial pellet was triturated under liquid nitrogen using a mortar and pestle and lysed at 277 K in a buffer consisting of 1 mg ml−1 lysozyme in 10 mM HEPES pH 8.0 containing 300 mM NaCl, 25 mM imidazole pH 8.0, 10 mM β-mercaptoethanol, 0.1 mM EDTA and protease inhibitors (0.2 mM PMSF, 0.1 mM TPCK, 7.6 µM leupeptin and 0.15 µM soybean trypsin inhibitor) (buffer A). After ∼30 min, the viscocity of the lysate was elevated, indicating that lysis was complete. DNase I and MgCl2 were then added to 20 µg ml−1 and 4 mM, respectively, and the mixture was stirred at room temperature until the viscosity of the lysate decreased (∼10 min). The lysate was centrifuged at 150 000g for 40 min and the resulting supernatant was collected. The supernatant was diluted twofold with buffer A lacking EDTA and protease inhibitors (buffer B) and was applied onto a 5 ml Ni2+–NTA Superflow column (Qiagen, Valencia, CA, USA) that was pre-equilibrated with buffer B. The column was washed with ten column volumes (50 ml) of buffer B and the protein was eluted with buffer C (buffer B containing 250 mM imidazole pH 8.0). 0.5 ml fractions were collected and those containing protein were identified using a modified version of the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Protein-containing fractions were pooled, TEV protease (Kapust et al., 2001 ▶) was added to a concentration of 3%(w/w) (based on protein concentration) and the mixture was dialyzed against 4 l buffer B overnight. The dialyzed protein solution was again applied onto a pre-equilibrated Ni2+–NTA column in order to remove the cleaved MBP-His10 tag, uncleaved fusion protein and TEV protease, all of which contain His6 or His10 tags, and the flowthrough containing cut fusion protein was collected in 0.5 ml fractions. Protein-containing fractions were again identified using the modified Bio-Rad assay and subjected to further analysis via SDS–PAGE. After the second Ni2+–NTA affinity purification, a prominent ∼60 kDa band was observed on a Coomassie-stained SDS–PAGE gel. Fractions containing this ∼60 kDa band were pooled and the protein was concentrated to ∼8 mg ml−1 using a 50 kDa MWCO Centricon (Millipore, Billerica, MA, USA). The buffer containing the concentrated protein consisted of 10 mM HEPES pH 8.0 containing 300 mM NaCl, 25 mM imidazole pH 8.0 and 10 mM β-mercaptoethanol. Crystallization conditions were tested in VDX plates (Hampton Research, Aliso Viejo, CA, USA) using the hanging-drop method and several commercial sparse-matrix screens by mixing 1 µl each of the protein and crystallization solutions and incubating the drops at 277 K with 1 ml reservoir solution. Thin bar-shaped crystals were observed after 3 d in condition No. 4 [0.1 M imidazole pH 8.0 containing 35%(v/v) 2-methyl-2,4-pentanediol and 0.2 M MgCl2] of the Wizard I screen (Emerald BioSystems, Bainbridge Island, WA, USA) (Fig. 1 ▶). Several crystals were extensively washed with well solution and dissolved in water for mass-spectrometry analysis. The protein constituting the crystals was identified as GroEL using MALDI and LC-MS/MS analysis. No components of the fusion protein were detected in the crystal by mass spectrometry and an SDS–PAGE gel of washed crystals revealed one ∼60 kDa band corresponding to GroEL. Optimization of crystallization conditions was achieved by increasing the MgCl2 concentration to 0.32 M, reducing the MPD concentration to 34% and increasing the total drop volume to 4 µl. These conditions yielded larger more single-looking crystals of maximum dimensions 1.0 × 0.4 × 0.05 mm that grew vertically in both hanging and sitting drops and diffracted to ∼2.6 Å using synchrotron radiation. Crystals were harvested using cryoloops (Hampton Research, Aliso Viejo, CA, USA) or litholoops (Molecular Dimensions, Apopka, FL, USA), were cryoprotected by soaking in well solution for 10 s and were flash-cooled by plunging into liquid nitrogen.
Figure 1.

Photograph of a GroEL crystal illuminated with polarized light. The crystals in this photograph grew to final size within one week at 277 K in a crystallization solution consisting of 100 mM imidazole pH 8 containing 34%(v/v) MPD and 0.32 M MgCl2. The scale bar represents ∼200 µm.
Additional experiments revealed that GroEL copurified with the same fusion protein when amylose resin was used in place of Ni2+–NTA for affinity chromatography. Furthermore, we observed that GroEL copurified with RPE65 containing N- or C-terminal six-histidine tags without fused MBP from Ni2+–NTA resin. Treatments that were designed to disrupt the interaction such as high-salt or detergent washes or buffers containing ATP, magnesium and potassium were all unsuccessful at completely dissociating GroEL from the fusion protein.
2.2. Data collection, structure determination and refinement
Diffraction intensities were collected at Advanced Light Source beamline 5.0.2. A data set consisting of 200 frames was collected using a wavelength of 0.97 Å, a crystal-to-detector distance of 400 mm, an oscillation angle of 1° and an exposure time of 10 s per frame. The data were indexed, integrated and scaled using the HKL-2000 software suite (Otwinowski & Minor, 1997 ▶) and the structure was solved by molecular replacement using the program Phaser (McCoy et al., 2005 ▶) with a previously determined GroEL structure (PDB code 1kp8) as a search model. The structure was refined using the program REFMAC (Murshudov et al., 1997 ▶) alternating with manual model fitting using the programs O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). Because of the sevenfold symmetry present in each of the GroEL rings, noncrystallographic symmetry (NCS) restraints (Table 1 ▶) were utilized and proved to be beneficial during the refinement process, ultimately leading to lower R cryst and R free values. Weak electron density was observed for the region comprised of residues 474–488, indicating a substantial degree of conformational variability; therefore, this region was not subjected to NCS restaints. Inclusion of translation–libration–screw (TLS) groups to model large-scale thermal motions also resulted in significant improvements in R cryst and R free and helped to elucidate regions with weak electron density (Winn et al., 2001 ▶). Three TLS groups per subunit, identical to those used by Chaudhry and coworkers in their refinement of a previously determined GroEL structure (PDB code 1oel) and corresponding to the apical, intermediate and equatorial domains, were used during the refinement (Table 1 ▶) (Chaudhry et al., 2004 ▶). Data-collection and refinement statistics are summarized in Table 2 ▶.
Table 1. NCS and TLS groups used during refinement.
Owing to the possibility of asymmetry between rings, NCS restraints were applied only within heptameric rings, not between rings. The NCS groups and levels of restraints were adjusted during the refinement process to improve the fit of the model to electron density. Each GroEL subunit was divided into three TLS groups and a total of 42 groups were used. The TLS groups are the same as those described previously (Chaudhry et al., 2004 ▶).
| NCS groups | |
| Tight restraints | Residues 139, 47195, 489525 |
| Loose restraints | Residues 4046, 196473 |
| TLS groups | |
| Group 1 | Residues 2135, 410525 |
| Group 2 | Residues 136190, 375409 |
| Group 3 | Residues 191374 |
Table 2. Data-collection and refinement statistics.
| Data collection | |
| Beamline | ALS 5.0.2 |
| Detector | ADSC Q315 |
| Wavelength () | 0.97 |
| Temperature (K) | 100 |
| Space group | P21 |
| Unit-cell parameters (, ) | a = 136.4, b = 262.3, c = 147.1, = 99.83 |
| Resolution range† () | 503.02 (3.133.02) |
| Total No. of observations | 759112 |
| No. of unique observations† | 202060 (20078) |
| Mosaicity () | 0.553 |
| Average redundancy† | 3.8 (3.6) |
| Completeness† (%) | 99.7 (98.9) |
| I/(I)† | 9.8 (1.02) |
| R sym † ‡ (%) | 8.6 (75.7) |
| Solvent content (%) | 63 |
| Refinement | |
| Refinement resolution† | 303.02 (3.0983.02) |
| R cryst § (%) | 22.7 (24.9) |
| R free § ¶ (%) | 26.3 (28.5) |
| No. of protein atoms | 54176 |
| Average B factor | 70.4 |
| R.m.s.d. for bond lengths () | 0.006 |
| R.m.s.d. for bond angles () | 0.971 |
| Ramachandran plot | |
| Most favored regions (%) | 93.7 |
| Additionally allowed regions (%) | 5.9 |
| Generously allowed regions (%) | 0.4 |
| Disallowed region (%) | 0 |
Values in parentheses are for the highest resolution shell.
R
sym = 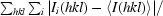
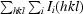 , where the summation is over all symmetry-equivalent reflections, excluding reflections observed only once.
, where the summation is over all symmetry-equivalent reflections, excluding reflections observed only once.
Values in parentheses are when TLS was omitted from the refinement.
R
free = 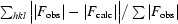 . R
free was calculated using a randomly selected 5.1% of the data.
. R
free was calculated using a randomly selected 5.1% of the data.
3. Results and discussion
In general, the crystal structure reported here is in agreement with the previously determined wild-type GroEL structure (Bartolucci et al., 2005 ▶). In contrast to the conditions previously used to crystallize wild-type GroEL, potassium salt was not included in either the purification or the crystallization solutions and no electron density is observed in the potassium ion-binding site between Leu31 and Lys51 in this structure.
In order to assess the differences between the two wild-type GroEL structures, we performed pairwise comparisons of every subunit of this structure with those of the previously determined wild-type GroEL structure (PDB code 1xck) as well as comparisons of subunits within each structure using Cruickshank’s diffraction precision index (Cruickshank, 1999 ▶) with the addition of linear B-factor scaling as implemented in the program ESCET (Schneider, 2000 ▶, 2002 ▶). In 75% (68/91) of the comparisons of subunits within our structure the subunits were found to be conformationally invariant, whereas in the other wild-type structure (1xck) 57% (52/91) of comparisons exhibited conformational invariability. In the pairwise comparisons between subunits in 1xck and our structure, in 123/196 (63%) no conformational variation was found. In these pairwise comparisons between the two wild-type structures, the regions which were found to be flexible included residues 44–45 located in the stem loop, residues 202–204 and 260–268 located in the apical domain and residues 477–487 located in the equatorial domain near the ATP-binding site (Fig. 2 ▶). All of these regions, with the exception of residues 477–487, have high temperature factors in other GroEL crystal structures. It is likely that the differences observed between 1xck and the current structure arise from the inherent thermal variability of these regions. The electron density associated with residues 477–487 was weak and the B factors were significantly higher in this region compared with previously determined GroEL structures (see below); therefore, it is possible that the differences between structures in this region arise from this ambiguity. In general, there were no regions that were consistently different between the current structure and 1xck, including those that constitute the potassium ion-binding site.
Figure 2.

Image of a GroEL monomer showing the location of the potassium ion-binding site as well as the regions that were identified as being most flexible in a comparison to the previously determined wild-type GroEL structure (PDB code 1xck). Residues 44–45, 202–204 and 260–268 line the folding cavity, while residues 477–487 are located on the outside surface near the ATP-binding site and ring–ring interface of the GroEL complex. This figure was generated using PyMOL v.0.99 (DeLano, 2002 ▶).
The thermal motions of the GroEL domains are well described through the use of TLS refinement (Chaudhry et al., 2004 ▶). Indeed, this was also the case with the current structure: a drop in R cryst and R free from 24.9 to 22.7 and from 28.5 to 26.3, respectively, was observed when TLS refinement was included in REFMAC. Furthermore, the inclusion of TLS parameters improved electron density in regions that were poorly ordered. Analysis of the residual isotropic temperature factors after TLS refinement indicated that the motion of only one loop, comprised of residues 474–488, was not well modeled by the TLS groups. REFMAC refinement of the structure without TLS parameters showed that the temperature factors associated with this loop were significantly higher in this structure compared with previously determined GroEL structures (both nucleotide-bound and apo forms) and the electron density in this area was weak and discontinuous. Electron-density maps were calculated after several rounds of refinement with loop 474–488 omitted from every subunit and the resulting 2F o − F c and F o − F c maps showed discontinuous density in this region for most subunits. Based on other GroEL crystal structures with bound nucleotide, a portion of this loop makes contacts with the adenyl moiety of ATP/ADP (Xu et al., 1997 ▶; Wang & Boisvert, 2003 ▶). Additionally, the loop is adjacent to a major inter-ring contact point. It is conceivable that ATP or ADP binding may stabilize this loop, resulting in conformational changes that affect the opposite ring.
4. Summary
In summary, we present a 3.02 Å crystal structure of wild-type apo GroEL purified from a natural source and crystallized in the absence of potassium ions. The observations made regarding the copurification of GroEL may have implications for structural genomics studies, which commonly use MBP fusions or other solubility-enhancing proteins for bacterial expression as a strategy to increase solubility as well as to aid in purification. It is possible that GroEL copurification may be a common event when using solubility-enhancing fusion proteins. Indeed, GroEL copurification with fusion proteins has been documented in the literature (Couch et al., 2002 ▶; Chen et al., 2001 ▶; Huang & Chuang, 1999 ▶).
Although it is generally accepted that the GroEL–GroES complex cannot encapsulate unfolded substrates with molecular weights above ∼60 kDa, there have been reports of GroEL promoting folding of substrate proteins above this molecular weight either through noncanonical binding or by GroEL adopting expanded conformations (Huang & Chuang, 1999 ▶; Chaudhuri et al., 2001 ▶; Chen et al., 2006 ▶). Our fusion protein bound GroEL quite tenaciously despite the fact that it is has a molecular weight greater than 100 kDa. It is likely that the fusion protein was only partially encapsulated by GroEL.
The overall structure is in agreement with previously determined apo GroEL structures. However, there are some differences with respect to temperature-factor value distribution. Additionally, potassium ions are not observed in the equatorial domain potassium ion-binding sites, which differs from the previously determined wild-type apo GroEL structure. However, the absence of potassium ions does not seem to cause any major structural changes, at least within the resolution of the structure.
Supplementary Material
PDB reference: GroEL, 2nwc, r2nwcsf
Acknowledgments
This research was supported in part by grant No. EY009339 from the National Eye Institute, National Institutes of Health (KP). PDK and DTL are supported by the Visual Sciences Training Program grant No. 2T32EY007157 from the National Eye Institute, National Institutes of Health. PDK is supported by the Molecular Therapeutics Training Program grant 5T32GM008803 from the National Institute of General Medical Sciences, National Institutes of Health. We would like to thank the staff at the ALS 5.0.2 and APS 19-ID beamlines for technical assistance, as well as the proteomics core facility at CWRU for assistance with mass-spectrometry data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC for the US Department of Energy under contract DE-AC02-06CH11357.
References
- Bartolucci, C., Lamba, D., Grazulis, S., Manakova, E. & Heumann, H. (2005). J. Mol. Biol. 354, 940–951. [DOI] [PubMed] [Google Scholar]
- Boisvert, D. C., Wang, J., Otwinowski, Z., Horwich, A. L. & Sigler, P. B. (1996). Nature Struct. Biol. 3, 170–177. [DOI] [PubMed] [Google Scholar]
- Braig, K., Adams, P. D. & Brünger, A. T. (1995). Nature Struct. Biol. 2, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D. C., Joachimiak, A., Horwich, A. L. & Sigler, P. B. (1994). Nature (London), 371, 578–586. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar, G. N., Tilly, K., Woolford, C., Hendrix, R. & Georgopoulos, C. (1986). J. Biol. Chem. 261, 12414–12419. [PubMed] [Google Scholar]
- Chaudhry, C., Horwich, A. L., Brunger, A. T. & Adams, P. D. (2004). J. Mol. Biol. 342, 229–245. [DOI] [PubMed] [Google Scholar]
- Chaudhuri, T. K., Farr, G. W., Fenton, W. A., Rospert, S. & Horwich, A. L. (2001). Cell, 107, 235–246. [DOI] [PubMed] [Google Scholar]
- Chen, D. H., Song, J. L., Chuang, D. T., Chiu, W. & Ludtke, S. J. (2006). Structure, 14, 1711–1722. [DOI] [PubMed] [Google Scholar]
- Chen, L. & Sigler, P. B. (1999). Cell, 99, 757–768. [DOI] [PubMed] [Google Scholar]
- Chen, X. S., Casini, G., Harrison, S. C. & Garcea, R. L. (2001). J. Mol. Biol. 307, 173–182. [DOI] [PubMed] [Google Scholar]
- Couch, R., Seidle, H. & Parry, R. (2002). Biotechniques, 32, 1230–1236. [DOI] [PubMed] [Google Scholar]
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fenton, W. A. & Horwich, A. L. (1997). Protein Sci. 6, 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houry, W. A., Frishman, D., Eckerskorn, C., Lottspeich, F. & Hartl, F. U. (1999). Nature (London), 402, 147–154. [DOI] [PubMed] [Google Scholar]
- Huang, Y. S. & Chuang, D. T. (1999). J. Biol. Chem. 274, 10405–10412. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kapust, R. B., Tozser, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D. & Waugh, D. S. (2001). Protein Eng. 14, 993–1000. [DOI] [PubMed] [Google Scholar]
- Kristelly, R., Earnest, B. T., Krishnamoorthy, L. & Tesmer, J. J. (2003). Acta Cryst. D59, 1859–1862. [DOI] [PubMed] [Google Scholar]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Ranson, N. A., Farr, G. W., Roseman, A. M., Gowen, B., Fenton, W. A., Horwich, A. L. & Saibil, H. R. (2001). Cell, 107, 869–879. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. (2000). Acta Cryst. D56, 714–721. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. (2002). Acta Cryst. D58, 195–208. [DOI] [PubMed] [Google Scholar]
- Todd, M. J., Viitanen, P. V. & Lorimer, G. H. (1993). Biochemistry, 32, 8560–8567. [DOI] [PubMed] [Google Scholar]
- Viitanen, P. V., Lubben, T. H., Reed, J., Goloubinoff, P., O’Keefe, D. P. & Lorimer, G. H. (1990). Biochemistry, 29, 5665–5671. [DOI] [PubMed] [Google Scholar]
- Wang, J. & Boisvert, D. C. (2003). J. Mol. Biol. 327, 843–855. [DOI] [PubMed] [Google Scholar]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed] [Google Scholar]
- Xu, Z., Horwich, A. L. & Sigler, P. B. (1997). Nature (London), 388, 741–750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: GroEL, 2nwc, r2nwcsf