

The crystallization of branched-chain aminotransferase from D. radiodurans is described.
Keywords: branched-chain amino-acid aminotransferase, Deinococcus radiodurans
Abstract
The branched-chain amino-acid aminotransferase (BCAT), which requires pyridoxal 5′-phosphate (PLP) as a cofactor, is a key enzyme in the biosynthetic pathway of the hydrophobic amino acids leucine, isoleucine and valine. DrBCAT from Deinococcus radiodurans, which has a molecular weight of 40.9 kDa, was crystallized using the hanging-drop vapour-diffusion method. According to X-ray diffraction data to 2.50 Å resolution from a DrBCAT crystal, the crystal belongs to space group P212121, with unit-cell parameters a = 56.37, b = 90.70, c = 155.47 Å. Preliminary analysis indicates the presence of two DrBCAT molecules in the asymmetric unit, with a solvent content of 47.52%.
1. Introduction
Branched-chain aminotransferases (BCATs; EC 2.6.2.42), which require pyridoxal 5′-phosphate (PLP) as a cofactor, reversibly catalyze the transfer of the α-amino group of the three most hydrophobic branched-chain amino acids (BCAAs), i.e. leucine, isoleucine and valine, to α-ketoglutarate to form the respective branched-chain α-keto acids and glutamate (Taylor & Jenkins, 1966 ▶; Ichihara & Koyama, 1966 ▶). The enzyme functions via a ping-pong kinetic mechanism that comprises two half-reactions each with three main steps (Kirsch et al., 1984 ▶). BCAT enzymes are distributed widely in many species (Ichihara, 1985 ▶; Conway & Hutson, 2000 ▶), from bacteria, which contain a single BCAT enzyme (Kamitori et al., 1989 ▶), to mammals, which have the enzyme in a mitochondrial and a cytosolic form (Hutson, 1988 ▶; Hutson et al., 1988 ▶). Of the four classified families of PLP-dependent enzymes with various fold types, the BCATs belong to the fold type IV family that transfers protons on the Re face of the PLP cofactor (Yoshimura et al., 1993 ▶; Jhee et al., 2000 ▶). BCAT can be used to synthesize l-tert-leucine and other branched-chain or unnatural amino acids from their respective keto acids; they have broad applicability in the synthesis of fine chemicals and pharmaceuticals (Taylor et al., 1998 ▶).
Deinococcus radiodurans R1 is an extremophile, red-pigmented, Gram-positive, nonmotile bacterium that is well known for its extreme resistance to the lethal effects of ionizing radiation, ultraviolet (UV) radiation and hydrogen peroxide (Minton, 1994 ▶; White et al., 1999 ▶). It was originally identified in irradiated canned meat and has been found widely in organic nutrients, including soil, animal faeces and processed meats, and in dry nutrient-poor environments, including weathered granite in a dry Antarctic valley, room dust and irradiated medical instruments (Masters et al., 1991 ▶). The complete genome sequence of D. radiodurans R1 has been determined (Lin et al., 1999 ▶; White et al., 1999 ▶); it reveals the related chromosomes and megaplasmid of a bacterium that is able to survive under conditions of starvation, oxidative stress and extensive DNA damage. The mechanism of repair of DNA double-strand breaks has received intensive study (Frenkiel-Krispin & Minsky, 2006 ▶; Minsky, 2003 ▶; Levin-Zaidman et al., 2003 ▶; Englander et al., 2004 ▶). The protein components of D. radiodurans are also valuable sources for investigating structure–function relationships owing to their possibly unique structures that might confer special properties such as thermostability or radioresistance.
The DrBCAT (BCAT from D. radiodurans) gene was cloned and expressed in Escherichia coli. The enzyme is expected to have special activity and stability in an environment of ionizing radiation in terms of structure and molecular packing. Here, we describe the successful purification, crystallization and X-ray crystallographic characterization of this 358 amino-acid enzyme, the first reported BCAT from a radioresistant organism.
2. Materials and methods
2.1. Protein purification and identification
To amplify the DrBCAT coding sequence by polymerase chain reaction (PCR), we used cDNA that had been prepared from genomic DNA of D. radiodurans as the template; the forward primer was 5′-GCGGATCCATGCGCCTTACAATC-3′ and the reverse primer was 5′-CCCAAGCTTCTACACCTTCACAATCCA-3′. The PCR products were cloned into the pQE30 vector (Qiagen) and tested by nucleotide sequencing. The reaction mixture was then transformed into E. coli JM109 and incubated on LB agar plates containing ampicillin (50 µg ml−1) at 310 K for 16 h. The DrBCAT clones were picked and their plasmids were screened by gel electrophoresis with agarose (1.2%) after a restriction cut with BamHI and HindIII. The putative plasmid containing the DrBCAT fragment was then further confirmed by nucleotide sequencing.
For the expression of DrBCAT, one colony of the bacteria was picked and placed into 200 ml Luria–Bertani (LB) broth containing 50 µg ml−1 ampicillin for incubation overnight until the OD600 value exceeded 1.0. The overnight culture was then transferred into six flasks each containing 1.5 l fresh LB medium with 100 µg ml−1 ampicillin. The culture was grown at 310 K with shaking at medium speed (140 rev min−1) until the OD600 reached 0.5–0.8 in 3 h. IPTG (final concentration 1 mM) was then added to the culture for induction and incubation was continued at 293 K for another 18 h. The whole cells were then harvested by centrifugation (6000 rev min−1) at 277 K for 30 min. The medium was discarded and the cell pellet was resuspended in 250 ml binding buffer containing 0.5 M NaCl, 5 mM imidazole and 20 mM Tris–HCl pH 7.9 and then subjected to cell disruption by ultrasonication and directed through a high-pressure homogenizer (138 MPa, four cycles; EmulsiFlex-C3, Avetin Inc., Canada). The suspension was collected by centrifugation (16 200 rev min−1) at 277 K for 30 min. The soluble protein extract was then passed through a His-tag Ni2+–NTA column (length 10 cm; General Electric Co.) that had been pre-equilibrated with 50 ml binding buffer. The column was washed with 200 ml binding buffer containing 20 mM imidazole; the purified protein was then eluted with 100 ml buffer containing 200 mM imidazole. The affinity chromatography on the Ni2+–NTA column was repeated to increase the homogeneity. The protein sample was concentrated using an Amicon (MWCO 10 000, Millipore) and the buffer was concurrently changed to 20 mM Tris buffer pH 7.0. The yield of the protein was approximately 7 mg per litre; the purity was better than 95% as analysed by SDS–PAGE (12.5%; Fig. 1 ▶) and UV–visible spectroscopy.
Figure 1.

Coomassie blue stained 12.5% Tricine SDS–PAGE of pools from DrBCAT purification. Purification fractions containing BCAT activity were collected at each step. All samples were boiled at 373 K for 10 min in 4× sample buffer. Lane 1, pool from total cell lysate in 0.3 M NaCl, 10 mM imidazole and 50 mM NaH2PO4 pH 8.0; lane 2, pool from supernatant after centrifugation; lane 3, pool from pellet after centrifugation; lane 4, pool from the first His-tag Ni2+–NTA column; lane 5, pool from the second His-tag Ni2+–NTA column; lane M, molecular-weight markers (kDa).
2.2. Crystallization
Prior to crystallization trials, the purified protein sample was prepared at a concentration of 14 mg ml−1 in 50 mM HEPES buffer pH 7.4 containing 10 µM pyridoxal phosphate (PLP). Crystallization was performed in VDX24 plates (Hampton Research) using the hanging-drop vapour-diffusion method at 291 K. Small crystals were observed from a condition consisting of 20%(w/v) PEG 8000 and 0.2 M magnesium chloride in 0.1 M Tris buffer pH 8.5 (Wizard II screen No. 3; Jena Bioscience) within 5 d of setup. This condition was refined further to produce larger BCAT crystals using 2 µl hanging drops containing equal volumes of protein solution (1 µl) and reservoir solution (1 µl) equilibrated against 200 µl reservoir solution containing 15%(w/v) PEG 8000 and 0.15 M MgCl2 in 0.1 M Tris buffer pH 7.3. Crystals of good diffraction quality were used for data collection (Fig. 2 ▶).
Figure 2.

Single needle crystals of DrBCAT grown by the hanging-drop method.
2.3. X-ray data collection and processing
The protein crystals were initially tested and characterized using synchrotron radiation at the SPXF beamlines BL13B1 and BL13C1 equipped with CCD detectors (Q315 and Q210, ADSC) at the National Synchrotron Radiation Research Center (NSRRC), Taiwan. Data collection was completed at the Taiwan-contracted protein crystallographic beamline BL12B2 equipped with a CCD detector (Quantum-4R, ADSC) at SPring-8 in Japan. The crystal was transferred from a crystallization drop into 5 µl cryoprotectant solution containing 15%(w/v) PEG 8000, 0.15 M MgCl2 and 20%(v/v) glycerol in 0.1 M Tris buffer pH 7.3 for a few seconds, mounted on a synthetic nylon loop (0.1–0.2 mm, Hampton Research) and then flash-cooled in liquid nitrogen at 100 K. For complete data collection, a 165° rotation was measured with 0.5° oscillations using an X-ray wavelength of 1.00 Å, an exposure time of 30 s and a crystal-to-detector distance of 200 mm at 100 K in a nitrogen stream using a cryo-system (X-Stream, Rigaku/MSC Inc.). All data were indexed, integrated and scaled using programs from the HKL-2000 suite (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Under SDS-denaturating and reducing conditions, SDS–PAGE showed a single band corresponding to a molecular weight of about 41 000 Da (Fig. 1 ▶), which is in agreement with the molecular weight of BCAT determined by ESI-tandem MS of 40 891 Da. Yellowish needle-shaped protein crystals appeared after 5 d and continued to grow to final dimensions of 0.5 × 0.02 × 0.02 mm after two weeks in an incubator at 291 K (Fig. 2 ▶). Larger crystals of rectangular shape occasionally also grew in the same crystallization drops, but diffracted poorly in terms of resolution. The protein crystals were sensitive to the change in precipitant concentration on being transferred to the cryoprotectant solution containing 20% glycerol. Crystals of satisfactory quality were identified by careful screening and selection for data collection, as they typically exhibited fairly high mosaicity (>1°). Radiation damage was observed after protracted exposure during data collection, which caused a decrease in I/σ(I) and an increase in R sym. Thus, although data for a total rotation of 180° were collected, only the first 165° was selected for data processing after an inspection of data statistics with regard to crystal decay. Analysis of the diffraction pattern indicated that these crystals exhibit orthorhombic symmetry and systematic absences indicated the space group to be P212121. Assuming the presence of two BCAT molecules per asymmetric unit, the Matthews coefficient is estimated to be 2.43 Å3 Da−1, corresponding to a solvent content of 47.52% (Matthews, 1968 ▶), which is within the general range for protein crystals. Details of data statistics are provided in Table 1 ▶.
Table 1. Diffraction statistics of DrBCAT.
Values in parentheses are for the highest resolution shell (2.50–2.41 Å).
| Wavelength (Å) | 1.00 |
| Temperature (K) | 100 |
| Resolution range (Å) | 30.0–2.5 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 56.37, b = 90.70, c = 155.47 |
| Unique reflections | 28352 |
| Completeness (%) | 98.6 (100) |
| 〈I/σ(I)〉 | 34.8 (4.5) |
| Average redundancy | 6.2 |
| Rsym† (%) | 3.9 (26.9) |
| Mosaicity (°) | 0.41 |
| No. of molecules per ASU | 2 |
| Matthews coefficient (Å3 Da−1) | 2.43 |
| Solvent content (%) | 47.52 |
R
sym = 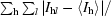
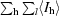 , where I
l is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
, where I
l is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
Initial attempts to solve the crystal structure of BCAT were performed by molecular replacement using the structure of human BCAT (33% sequence identity with 17 gaps; PDB code 2coj; Goto et al., 2005 ▶), with nonconserved residues altered to alanines, as a search model. A molecular-replacement solution was obtained with a correlation coefficient of 32.2 and an R factor of 47.4% (the next highest solution had a correlation coefficient of 24.0 and an R factor of 52.8%) in the resolution range 15–3.5 Å using CNS v.1.1 (Brünger et al., 1998 ▶), which confirmed the presence of two protein molecules per asymmetric unit. After rigid-body refinement using CNS v.1.1 (Brünger et al., 1998 ▶) in the resolution range 25–3.0 Å, the R and R free factors were 48.5% and 47.9%, respectively. Complete model building and refinement of the structure to 2.50 Å resolution is in progress; structural details will be described in a separate paper.
Acknowledgments
We are grateful to our colleagues Drs Yuch-Cheng Jean and Chun-Shiun Chao and supporting staff for technical assistance at the synchrotron-radiation X-ray facility during data collection at BL13B1 and BL13C1 of NSRRC in Taiwan and Yu-San Huang, Kuan-Li Yu and Tzu-Ling Lee at BL12B2 of SPring-8 in Japan. This study was supported in part by National Synchrotron Radiation Research Center 944RSB02 and National Science Council NSC 94-2321-B-213-001 in Taiwan to C-JC.
References
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Conway, M. E. & Hutson, S. M. (2000). Methods Enzymol.324, 355–365. [DOI] [PubMed] [Google Scholar]
- Englander, J., Klein, E., Brumfeld, V., Sharma, A. K., Doherty, A. J. & Minsky, A. (2004). J. Bacteriol.186, 5973–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkiel-Krispin, D. & Minsky, A. (2006). J. Struct. Biol.156, 311–319. [DOI] [PubMed] [Google Scholar]
- Goto, M., Miyahara, I., Hirotsu, K., Conway, M., Yennawar, N., Islam, M. M. & Hutson, S. M. (2005). J. Biol. Chem.280, 37246–37256. [DOI] [PubMed] [Google Scholar]
- Hutson, S. M. (1988). J. Nutr.118, 1475–1481. [DOI] [PubMed] [Google Scholar]
- Hutson, S. M., Fenstermacher, D. & Mahar, C. (1988). J. Biol. Chem.263, 3618–3625. [PubMed] [Google Scholar]
- Ichihara, A. (1985). Transaminases, edited by P. Christen & D. E. Metzler, Vol. 2, pp. 430–439. New York: Wiley.
- Ichihara, A. & Koyama, E. (1966). J. Biochem. (Tokyo), 59, 160–169. [DOI] [PubMed] [Google Scholar]
- Jhee, K.-H., Yoshimura, T., Miles, E., Takeda, S., Miyahara, I., Hirotsu, K., Soda, K., Kawata, Y. & Esaki, N. (2000). J. Biochem. (Tokyo), 128, 679–686. [DOI] [PubMed] [Google Scholar]
- Kamitori, S., Odagaki, Y., Inoue, K., Kuramitsu, S., Kagamiyama, H., Matsuura, Y. & Higuchi, T. (1989). J. Biochem. (Tokyo), 105, 671–672. [DOI] [PubMed] [Google Scholar]
- Kirsch, J. F., Eichele, G., Ford, G. C., Vincent, M. G., Jansonius, J. N., Gehring, H. & Christen, P. (1984). J. Mol. Biol.174, 497–525. [DOI] [PubMed] [Google Scholar]
- Levin-Zaidman, S., Englander, J., Shimoni, E., Sharma, A. K., Minton, K. W. & Minsky, A. (2003). Science, 299, 254–256. [DOI] [PubMed] [Google Scholar]
- Lin, J., Qi, R., Aston, C., Jing, J., Anantharaman, T. S., Mishra, B., White, O., Daly, M. J., Minton, K. W., Venter, J. C. & Schwartz, D. C. (1999). Science, 285, 1558–1562. [DOI] [PubMed] [Google Scholar]
- Masters, C. I., Murray, R. G., Moseley, B. E. & Minton, K. W. (1991). J. Gen. Microbiol.137, 1459–1469. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Minsky, A. (2003). Mol. Microbiol.50, 367–376. [DOI] [PubMed] [Google Scholar]
- Minton, K. W. (1994). Mol. Microbiol.13, 9–15. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Taylor, R. T. & Jenkins, W. T. (1966). J. Biol. Chem.241, 4396–4405. [PubMed] [Google Scholar]
- Taylor, P. P., Pantaleone, D. P., Senkpeil, R. F. & Fotheringham, I. G. (1998). Trends Biotechnol.16, 412–418. [DOI] [PubMed] [Google Scholar]
- White, O. et al. (1999). Science, 286, 1571–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, T., Nishimura, K., Ito, J., Esaki, N., Kagamiyama, H., Manning, J. & Soda, K. (1993). J. Am. Chem. Soc.115, 3898–3900.