

The crystallization of human complement regulator FH-678402H with a glycosaminoglycan analogue is described.
Keywords: complement factor H, age-related macular degeneration, complement regulator, short consensus repeat, sucrose octasulfate
Abstract
Human plasma protein complement factor H (FH) is an inhibitor of the spontaneously activated alternative complement pathway. An allotypic variant of FH, 402His, has been associated with age-related macular degeneration, the leading cause of blindness in the elderly. Crystals of FH domains 6–8 (FH678) containing 402His have been grown in the presence of a polyanionic sucrose octasulfate ligand (an analogue of the natural glycosaminoglycan ligands of FH) using both native and selenomethionine-derivatized protein. Native data sets diffracting to 2.3 Å and SeMet data sets of up to 2.8 Å resolution have been collected. An anomalous difference Patterson map reveals self- and cross-peaks from two incorporated Se atoms. The corresponding selenium substructure has been solved.
1. Introduction
Complement factor H (FH) is a fluid-phase regulator of the alternative pathway of the complement system. FH circulates in plasma and binds to polyanionic structures on self cells in order to inhibit complement attack propagated by the spontaneous activation of the alternative pathway (Fearon, 1978 ▶). Protection is provided by (i) the C3 convertase decay accelerating the activity of self-bound FH, which competes with the activator factor B for C3b binding, and (ii) FH acting as a cofactor in factor I-mediated cleavage of C3b (Whaley & Ruddy, 1976 ▶; Pangburn et al., 1977 ▶; Weiler et al., 1976 ▶). Both of these regulatory mechanisms reduce the amount of active C3 convertase and prevent further amplification of complement activity.
FH is a 150 kDa protein composed of 20 short consensus repeat (SCR) or complement control protein (CCP) domains. Each SCR is ∼60 amino acids long and contains two conserved disulfide bonds and a tryptophan, but SCR domains are otherwise diverse in sequence (Ripoche et al., 1988 ▶), allowing the various domains of FH to bind a wide range of ligands. FH binding sites for C3b (Jokiranta et al., 2000 ▶; Sharma & Pangburn, 1996 ▶) and heparin (Pangburn et al., 1991 ▶; Blackmore et al., 1996 ▶, 1998 ▶; Prodinger et al., 1998 ▶) have been mapped. FH is also a common point of interaction for microbial pathogens that evade complement attack. Borrelia burgdorferi (Kraiczy et al., 2001 ▶; Alitalo et al., 2001 ▶; Stevenson et al., 2002 ▶; Hellwage et al., 2001 ▶), Streptococcus pyogenes (Blackmore et al., 1998 ▶; Kotarsky et al., 1998 ▶) and Neisseria meningitidis (Ram et al., 1999 ▶) all have mechanisms for sequestering FH to their surfaces, allowing the bacteria to hijack the host self-defence mechanism against the alternative pathway and to evade complement attack.
An allotypic variant with histidine instead of tyrosine at position 402 in FH SCR7 has been identified as being associated with age-related macular degeneration (AMD; Klein et al., 2005 ▶; Edwards et al., 2005 ▶; Haines et al., 2005 ▶), the leading cause of blindness in adults in industrialized countries (Klein et al., 2002 ▶). Approximately 35% of people of European descent carry the 402His allele and being homozygous (10% of that population) for this disease-associated allele increases the risk of AMD by up to 7.4-fold (Klein et al., 2005 ▶).
To date, the solution structures of FH domains 5 (Barlow et al., 1992 ▶), 15–16 (Barlow et al., 1993 ▶), 16 (Norman et al., 1991 ▶) and 19–20 (Herbert et al., 2006 ▶) have all been solved by nuclear magnetic resonance, with a crystal structure of SCR pair 19–20 also having been determined more recently (Jokiranta et al., 2006 ▶). Here, we present details of the expression, purification, crystallization and preliminary crystallographic analysis of crystals of the FH domains 6–8 of the 402His variant (FH678402H) grown in the presence of sucrose octasulfate. Structural information about this region, which is critically implicated in disease, would significantly increase our understanding of this system.
2. Experimental procedures
2.1. Expression and purification
Generation of recombinant FH-678402H in a pET14b expression vector has been described previously (Clark et al., 2006 ▶). The protein was expressed in Escherichia coli BL21 (DE3) as inclusion bodies. The insoluble protein was then isolated from the cell lysate by centrifugation and washed in phosphate-buffered saline, before being completely denatured and reduced in solubilization buffer (0.1 M Tris pH 8, 8 M urea, 1 mM EDTA and 25 mM DTT). The protein was then refolded using a cysteine/cystine redox buffer, as described previously for the preparation of CD55 (White et al., 2004 ▶). Soluble protein was then purified by affinity chromatography using a 5 ml HiTrap Heparin column (GE Healthcare) and eluted using a gradient of 0.15–1.0 M NaCl, 50 mM Tris pH 7.5, 1 mM EDTA. This affinity step also serves as a test for the functionality of this region of FH, indicating correct refolding. Eluted protein was shown by SDS–PAGE analysis to be suitably pure for crystallization trials. This protein was dialysed overnight into 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA using SnakeSkin dialysis tubing (Perbio Science Europe) and concentrated to 2.6 mg ml−1 using Vivaspin 15 ml centrifugal concentrators (Sartorius) for storage at 193 K.
Selenomethionine derivatization of the protein was used to incorporate Se atoms into the structure. The FH-678402H-pET14b construct was purified from E. coli BL21 (DE3) using a QIAquick Miniprep kit (Qiagen) and transformed into E. coli B834 (DE3) methionine-auxotroph cells to ensure uniform selenomethionine labelling. Selenomethionine-derivatized protein was prepared in the same way as the native protein, except that the protein was expressed overnight at 294 K as opposed to 310 K. Inclusion bodies were isolated and the protein was refolded and purified as for the native protein.
2.2. Crystallization
All crystals were grown using the sitting-drop vapour-diffusion technique with initial crystallization conditions found by sparse-matrix screening (Jancarik & Kim, 1991 ▶). Drops contained 0.2 µl reservoir solution and 0.2 µl protein solution (2.6 mg ml−1, 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA with or without a tenfold molar excess of sucrose octasulfate ligand). Drops were equilibrated against 100 µl reservoir solution at 293 K. No leads were obtained in the absence of sucrose octasulfate; crystals only grew in the presence of sucrose octasulfate in condition No. 45 of Wizard Screen 1 (Emerald Biostructures) [0.1 M sodium acetate pH 4.5, 20%(w/v) PEG 3000]. No further optimization of this condition was required. Microseeding with crushed native crystals diluted 1:400 in reservoir solution was necessary to obtain crystals of SeMet FH-678402H that were large enough for the collection of diffraction data. The reservoir solution was the same as for the native crystals, except for dilution with 5%(v/v) glycerol. Bromide derivatives were prepared by soaking crystals of the native protein in a drop of reservoir solution saturated with NaBr for 20 s.
2.3. Data collection and processing
Native crystals (Fig. 1 ▶) were cryoprotected with reservoir solution diluted with 25%(v/v) glycerol and flash-frozen in liquid nitrogen. SeMet crystals were cryoprotected in the same way and NaBr-soaked crystals were protected using reservoir solution saturated with NaBr and 25%(v/v) glycerol.
Figure 1.

Native crystals of FH-678402H. Average crystal dimensions were approximately 200 × 50 × 5 µm.
Diffraction data sets were collected at 100 K at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). A fluorescence scan on an SeMet crystal (‘SeMet 2 peak’ data set) across the Se K edge gave values of 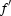 = −6.7 and
= −6.7 and 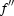 = 5.6 for the Se atoms in the crystal using the program CHOOCH (Evans & Pettifer, 2001 ▶). All crystals gave a diffraction pattern compatible with an orthorhombic C-centred lattice. All data were indexed and integrated using MOSFLM (Leslie, 1992 ▶) and scaled using SCALA (Evans, 1993 ▶) within the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). Processing statistics are given in Table 1 ▶. Systematic absences of the 00l reflections showed unambiguously that all crystals belonged to the C2221 space group.
= 5.6 for the Se atoms in the crystal using the program CHOOCH (Evans & Pettifer, 2001 ▶). All crystals gave a diffraction pattern compatible with an orthorhombic C-centred lattice. All data were indexed and integrated using MOSFLM (Leslie, 1992 ▶) and scaled using SCALA (Evans, 1993 ▶) within the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). Processing statistics are given in Table 1 ▶. Systematic absences of the 00l reflections showed unambiguously that all crystals belonged to the C2221 space group.
Table 1. FH-678402H diffraction data-collection statistics.
Values in parentheses correspond to the highest resolution shell.
| Data set | Native | SeMet 1 peak | SeMet 2 peak | SeMet 4 remote | Sulfur 1 | Sulfur 2 | NaBr soak 1 | NaBr soak 2 |
|---|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.978 | 0.979 | 0.979 | 0.975 | 1.20 | 0.815 | 0.920 | 0.920 |
| Space group | C2221 | C2221 | C2221 | C2221 | C2221 | C2221 | C2221 | C2221 |
| Unit-cell parameters | ||||||||
| a (Å) | 74.7 | 75.0 | 75.5 | 75.6 | 75.0 | 74.8 | 75.5 | 75.3 |
| b (Å) | 92.5 | 92.5 | 90.5 | 90.7 | 93.0 | 92.6 | 90.9 | 91.1 |
| c (Å) | 57.3 | 57.1 | 56.6 | 56.5 | 57.2 | 57.5 | 57.2 | 57.7 |
| α = β = γ (°) | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| Resolution (Å) | 20.0–2.35 (2.48–2.35) | 35.9–2.50 (2.64–2.50) | 31.4–2.9 (3.1–2.9) | 31.4–2.8 (3.0–2.8) | 46.5–2.9 (3.0–2.9) | 40.9–2.9 (3.1–2.9) | 31.5–3.10 (3.27–3.10) | 29.0–3.30 (3.48–3.30) |
| Completeness | 98.1 (88.9) | 99.4 (99.9) | 99.8 (100) | 99.9 (99.9) | 99.8 (98.3) | 100 (100) | 99.8 (99.9) | 99.6 (99.9) |
| Multiplicity | 6.8 (5.6) | 3.7 (3.8) | 7.7 (8.0) | 7.5 (7.7) | 11.0 (7.4) | 13.6 (14.4) | 5.3 (5.5) | 5.7 (6.0) |
| Unique reflections | 8392 (1075) | 7083 (1011) | 4526 (641) | 5030 (727) | 4224 (153) | 4676 (669) | 3788 (550) | 3163 (451) |
| Rmerge† (%) | 9 (42.7) | 8 (35.2) | 9.5 (30.3) | 9.3 (33.3) | 11.1 (34.3) | 8.7 (16.8) | 12.2 (38.5) | 1 (22.4) |
| I/σ(I) | 16.0 (4.4) | 12.5 (3.0) | 18.2 (6.0) | 17.7 (5.2) | 21.0 (5.2) | 25.5 (12.5) | 13.5 (5.5) | 16.3 (7.0) |
| Ranom‡ (%) | — | 5.3 (35) | 18 (35) | 18 (35) | 16 (34) | 15.3 (20) | 18.2 (67) | 29 (61) |
R
merge = 100 × 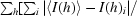
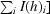 , where I(h)i is the ith observation of reflection h and 〈I(h)〉 is the mean intensity of all observations of h.
, where I(h)i is the ith observation of reflection h and 〈I(h)〉 is the mean intensity of all observations of h.
R
anom = 100 × 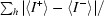
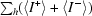 , where 〈I
+〉 and 〈I
−〉 are the mean intensities of the Bijvoet pairs for observation h.
, where 〈I
+〉 and 〈I
−〉 are the mean intensities of the Bijvoet pairs for observation h.
The selenium substructure of the SeMet crystals was investigated by generating an anomalous difference Patterson map (Fig. 2 ▶) using (E 2 − 1) coefficients and strict outlier rejection; the map was calculated using ECALC and FFT from CCP4 within the autoSHARP suite of programs (Vonrhein et al., 2006 ▶). The program SHELXD (Sheldrick & Schneider, 1997 ▶) within autoSHARP finds two Se sites, which were then refined using SHARP (de La Fortelle & Bricogne, 1997 ▶). Attempts at phasing using both SAD and MAD were also made using SHARP. Analysis of the log-likelihood gradient maps in SHARP revealed a 5.6σ peak in the anomalous difference data of NaBr soak 1.
Figure 2.

Harker section, u = 0.0, of the anomalous difference Patterson map of FH-678402HSe (data set SeMet 2 in Table 1 ▶). The Patterson map is calculated at 4 Å resolution. Maps are drawn with a minimum contour level of 1.5σ and 0.5σ increments.
3. Results and discussion
Native and selenomethionine-derivatized FH-678402H proteins crystallize as clusters of blades (Fig. 1 ▶). The Matthews coefficient is 2.36 Å3 Da−1 for one molecule in the asymmetric unit, corresponding to a solvent content of 47.9% (Matthews, 1968 ▶).
The anomalous difference Patterson maps calculated for the ‘SeMet 2 peak’ data set show anomalous signal peaks which can be explained by the two Se sites found by SHELXD/autoSHARP. The first of these two Se atoms gives a 4.5σ peak marked ‘A’ in Fig. 2 ▶. The second selenium gives a very weak anomalous signal and is marked ‘B’. Because the atoms share a common x coordinate, the cross-peaks between them are also visible in the plot and are labelled ‘C’. An additional peak at 3.1σ (labelled ‘D’ in Fig. 2 ▶) is yet to be explained and we assume that it may arise from errors in the highly anisotropic data. Owing to the weakness of the signal from the second selenium site, phasing with this two-Se-atom model has been unsuccessful to date. Alternative strategies are being sought to determine this structure.
Acknowledgments
We would like to thank the staff at the ESRF protein crystallography beamlines, Ed Lowe, Martin Noble and other members of The Laboratory of Molecular Biophysics, Oxford for assistance with data collection. Marc Morgan and Jenny Gibson maintained the crystallization robot in good order. BEP was funded by a studentship from the Wellcome Trust, SJ and PR by grants from the Medical Research Council.
References
- Alitalo, A., Meri, T., Rämö, L., Jokiranta, T. S., Heikkilä, T., Seppälä, I. J., Oksi, J., Viljanen, M. & Meri, S. (2001). Infect. Immun.69, 3685–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, P. N., Norman, D. G., Steinkasserer, A., Horne, T. J., Pearce, J., Driscoll, P. C., Sim, R. B. & Campbell, I. D. (1992). Biochemistry, 14, 3626–3634. [DOI] [PubMed]
- Barlow, P. N., Steinkasserer, A., Norman, D. G., Kieffer, B., Wiles, A. P., Sim, R. B. & Campbell, I. D. (1993). J. Mol. Biol.232, 268–284. [DOI] [PubMed] [Google Scholar]
- Blackmore, T. K., Fischetti, V. A., Sadlon, T. A., Ward, H. M. & Gordon, D. L. (1998). Infect. Immun.66, 1427–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore, T. K., Sadlon, T. A., Ward, H. M., Lublin, D. M. & Gordon, D. L. (1996). J. Immunol.157, 5422–5427. [PubMed] [Google Scholar]
- Clark, S. J., Higman, V. A., Mulloy, B., Perkins, S. J., Lea, S. M., Sim, R. B. & Day, A. J. (2006). J. Biol. Chem.281, 24713–24720. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Edwards, A. O., Ritter, R. III, Abel, K. J., Manning, A., Panhuysen, C. & Farrer, L. A. (2005). Science, 308, 421–424. [DOI] [PubMed] [Google Scholar]
- Evans, G. & Pettifer, R. (2001). J. Appl. Cryst.34, 82–86. [Google Scholar]
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Fearon, D. T. (1978). Proc. Natl Acad. Sci. USA, 75, 1971–1975. [Google Scholar]
- Haines, J. L., Hauser, M. A., Schmidt, S., Scott, W. K., Olson, L. M., Gallins, P., Spencer, K. L., Kwan, S. Y., Noureddine, M., Gilbert, J. R., Schnetz-Boutaud, N., Agarwal, A., Postel, E. A. & Pericak-Vance, M. A. (2005). Science, 308, 419–421. [DOI] [PubMed] [Google Scholar]
- Hellwage, J., Meri, T., Heikkila, T., Alitalo, A., Panelius, J., Lahdenne, P., Seppala, I. J. T. & Meri, S. (2001). J. Biol. Chem.276, 8427–8435. [DOI] [PubMed] [Google Scholar]
- Herbert, A. P., Uhrin, D., Lyon, M., Pangburn, M. K. & Barlow, P. N. (2006). J. Biol. Chem.281, 16512–16520. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Jokiranta, T. S., Hellwage, J., Koistinen, V., Zipfel, P. F. & Meri, S. (2000). J. Biol. Chem.275, 27657–27662. [DOI] [PubMed] [Google Scholar]
- Jokiranta, T. S., Jaakola, V., Lehtinen, L. J., Pärepalo, M., Meri, S. & Goldman, A. (2006). EMBO J.25, 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R., Klein, B. E. K., Tomany, S. C., Meuer, S. M. & Huang, G.-H. (2002). Ophthalmology, 109, 1767–1779. [DOI] [PubMed] [Google Scholar]
- Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J.-Y., Sackler, R. S., Haynes, C., Henning, A. K., SanGiovanni, J. P., Mane, S. M., Mayne, S. T., Bracken, M. B., Ferris, F. L., Ott, J., Barnstable, C. & Hoh, J. (2005). Science, 308, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotarsky, H., Hellwage, J., Johnsson, E., Skerka, C., Svensson, H. G., Lindahl, G., Sjobring, U. & Zipfel, P. F. (1998). J. Immunol.160, 3349–3354. [PubMed] [Google Scholar]
- Kraiczy, P., Skerka, C., Kirschfink, M., Brade, V. & Zipfel, P. F. (2001). Eur. J. Immunol.31, 1674–1684. [DOI] [PubMed] [Google Scholar]
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Norman, D. G., Barlow, P. N., Baron, M., Day, A. J., Sim, R. B. & Campbell, I. D. (1991). J. Mol. Biol.219, 717–725. [DOI] [PubMed] [Google Scholar]
- Pangburn, M. K., Atkinson, M. A. & Meri, S. (1991). J. Biol. Chem.266, 16847–16853. [PubMed] [Google Scholar]
- Pangburn, M. K., Schreiber, R. D. & Muller-Eberhard, H. J. (1977). J. Exp. Med.146, 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodinger, W. M., Hellwage, J., Spruth, M., Dierich, M. P. & Zipfel, P. F. (1998). Biochem. J.331, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram, S., Mackinnon, F. G., Gulati, S., McQuillen, D. P., Vogel, U., Frosch, M., Elkins, C., Guttormsen, H. K., Wetzler, L. M., Oppermann, M., Pangburn, M. K. & Rice, P. A. (1999). Mol. Immunol.36, 915–928. [DOI] [PubMed] [Google Scholar]
- Ripoche, J., Day, A. J., Harris, T. J. & Sim, R. B. (1988). Biochem. J.249, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. K. & Pangburn, M. K. (1996). Proc. Natl Acad. Sci. USA, 93, 10996–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick, G. & Schneider, T. (1997). Methods Enzymol.277, 319–343. [PubMed]
- Stevenson, B., El-Hage, N., Hines, M. A., Miller, J. C. & Babb, K. (2002). Infect. Immun.70, 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Methods Mol. Biol.364, 215–230. [DOI] [PubMed] [Google Scholar]
- Weiler, J. M., Daha, M. R., Austen, K. F. & Fearon, D. T. (1976). Proc. Natl Acad. Sci. USA, 73, 3268–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley, K. & Ruddy, S. (1976). J. Exp. Med.114, 1147–1163. [DOI] [PMC free article] [PubMed]
- White, J., Lukacik, P., Esser, D., Steward, M., Giddings, N., Bright, J. R., Fritchley, S. J., Morgan, B. P., Lea, S. M., Smith, G. P. & Smith, R. A. G. (2004). Protein Sci.13, 2406–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]