

The synthesis and crystallization of glucagon-Cex are reported.
Keywords: glucagon, exendin-4, diabetes, obesity
Abstract
Glucagon and glucagon-like peptide 1 (GLP-1) are drugs or drug candidates for the treatment of metabolic diseases such as diabetes and obesity. The native hormones have pharmacological deficiencies such as short half-life and poor solubility. A novel glucagon receptor agonist named glucagon-Cex has been designed, synthesized and crystallized. This peptide was highly soluble under physiological conditions and crystallized readily. The crystal diffracted X-rays to 2.2 Å resolution and the diffraction was consistent with space group P23, with unit-cell parameters a = b = c = 48.20 Å, α = β = γ = 90.0°. The crystals were suitable for a full structural determination to reveal the conformational differences between glucagon-Cex and the native hormone.
1. Introduction
Diabetes and obesity have become epidemic diseases and cause a significant burden to our healthcare system (Wyatt et al., 2006 ▶). Glucagon and GLP-1 analogs constitute an important family of drugs or drug candidates for the treatment of diabetes and obesity (Murphy et al., 2006 ▶). The pharmacological actions of this peptide family includes such central functions as glucose homeostasis, gastric emptying, intestinal growth and insulin secretion as well as the regulation of appetite (Larsen et al., 2003 ▶; Meier et al., 2002 ▶).
GLP-1 acts as an incretin hormone that stimulates insulin secretion in a glucose-dependent manner (Landsberg, 2006 ▶; Nauck et al., 1997 ▶; Nauck, 1998 ▶). Multiple clinical trials have demonstrated the efficacy of GLP-1 in controlling blood glucose, lowering HbA1c and restoring β-cell function. However, owing to the short biological half-life of native GLP-1, long-acting analogs are required for pharmaceutical intervention. A naturally occurring reptilian GLP-1 receptor agonist, exendin-4, has a nine-amino-acid C-terminal extension (Cex) and displays similar functional properties to native GLP-1 but with a much longer half-life (Doyle et al., 2003 ▶; Schepp et al., 1994 ▶). It has been developed as a new treatment for type II diabetes.
Glucagon is the most proven medication for treating acute hypoglycemia. While highly effective, its poor physical properties compromise formulation of the peptide since its solubility is extremely limited (Fanelli et al., 2006 ▶). Treatments for individuals with type II diabetes mostly involve regular insulin injections and constant management of their blood glucose levels. Even with constant monitoring, many people with diabetes mellitus are susceptible to acute hypoglycemia, a life-threatening condition in which glucose in the body drops to dangerously low levels. Administration of glucagon leads to a rapid rise in blood glucose. Glucagon is commercially supplied as a lyophilized powder to be solubilized in a dilute acid buffer and the solution immediately injected intramuscularly. While highly effective, the methodology is dangerous for a patient who is semi-conscious. Consequently, the biophysical properties of native glucagon are not conducive to a patient-friendly formulation and a more continently delivered glucagon agonist is needed (Bolli, 1984 ▶; Estall & Drucker, 2006 ▶; Gearhart & Parbhoo, 2006 ▶). A glucagon agonist with enhanced biophysical stability would present a leap forward in the control of insulin-dependent diabetes. In a search for more soluble and potent glucagon analogs for convenient therapeutic application, we synthesized a novel glucagon receptor agonist, glucagon-Cex, which consists of the native glucagon sequence elongated with the nine amino acids of Cex from exendin-4 (Fig. 1 ▶). This novel peptide exhibits greatly improved biophysical solubility and stability and hence represents a good drug candidate for the treatment of diabetes-related hypoglycemia. Here, we report the crystallization and preliminary crystallographic studies of this novel peptide hormone.
Figure 1.

Sequences of the peptides.
2. Materials and methods
2.1. Design and peptide synthesis
Exendin-4 is a peptide hormone originally extracted from the venom of the Heloderma lizard species that shares 53% amino-acid sequence similarity to human GLP-1. Exendin-4 has been shown to have a much longer half-life and higher agonistic activity towards GLP-1 receptor compared with native GLP-1 (Estall & Drucker, 2006 ▶; Fehse et al., 2005 ▶). One of the major differences between GLP-1 and exendin-4 is the addition of the nine Cex amino acids to the latter peptide. Studies have shown that the addition of Cex to GLP-1 also significantly increases the GLP-1 receptor activity (Goke et al., 1993 ▶). Therefore, the structural conformation of the carboxyl-terminal extension of these peptide hormones plays an important role in their biological function. It has been hypothesized that Cex forms a tryptophan cage for the C-terminal conserved hydrophobic residue (Neidigh et al., 2001 ▶). To explore the effect of Cex on glucagon, we designed the peptide glucagon-Cex.
Glucagon-Cex was synthesized on a solid support using Boc-based in situ neutralization chemistry on a highly modified Applied Biosystems 430A peptide synthesizer starting from 0.2 mmol methoxybenzhydrylamine resin (Schnolzer et al., 1992 ▶). The peptide was then cleaved from the support using HF/p-cresol in a 95:5 ratio for 1 h at 273 K. Following HF removal and ether precipitation, the peptide was extracted into 10% acetic acid and lyophilized. The peptide was then purified using RP-HPLC in 0.1% TFA using a linear gradient of acetonitrile on a Waters Associates preparative HPLC system. Fractions containing the desired peptide were pooled and lyophilized. The identity and purity of each analog was confirmed by analytical HPLC and MS analyses. The molecular weight of glucagon-Cex is 4316.7 Da. To facilitate the crystal structure determination, we also synthesized a selenomethionyl derivative of glucagon-Cex with an ABI-430A peptide synthesizer using Boc amino acids as described previously (Jin et al., 2000 ▶). The Boc seleno-l-methionine was prepared from l-selenomethionine using di-t-butyldicarbonate. The selenomethionyl glucagon-Cex was purified on a Kromasil C18 column using a gradient of 10–50% acetonitrile in 0.1% trifluoroacetic acid at pH 2 and repurified on a Phenomenex Primeshere 10 C18 column with a gradient of 15–35% acetonitrile in 0.05 M ammonium bicarbonate pH 8 on an FPLC system. Identified fractions were pooled, frozen and lyophilized. Mass-spectrometric analysis showed complete incorporation of selenomethionine into the peptide.
2.2. Crystallization
Lyophilized glucagon-Cex was solubilized in water at a concentration of 10 mg ml−1 prior to crystallization screening. Initial crystallization screening was performed using the sitting-drop vapor-diffusion method in 24-well plates. Equal volumes (1 µl) of the protein solution and reservoir solution were mixed. Each sitting drop was placed over 200–300 µl reservoir solution. If no promising crystal had formed in the drops after about 5 d, the plates were relocated from 298 to 277 K.
Hampton Research screening kits were used for initial crystallization attempts. To our surprise, more than ten conditions produced crystals readily. From the Index Screen, five different conditions produced crystals: (i) 1 M sodium acetate pH 4.5, 25%(w/v) PEG 3350, (ii) 0.1 M succinic acid pH 7.0, 15%(w/v) PEG 3350, (iii) 0.2 M magnesium chloride, 0.1 M HEPES pH 7.5, 25%(w/v) PEG 3350, (iv) 0.2 M proline, 0.1 M HEPES pH 7.5, 10%(w/v) PEG 3350 and (v) 0.1 M ammonium acetate, 0.1 M bis-Tris pH 5.5, 17%(w/v) PEG 10 000. From screening kit HR2-110, four conditions produced crystals: (i) 20% PEG 8000, 0.1 M sodium cacocylate pH 6.5, 0.2 M magnesium acetate, (ii) 30% PEG 8000, 0.2 M ammonium sulfate, (iii) ammonium sulfate and (iv) 8% PEG 8000, 0.1 M Tris–HCl pH 8.5. From screening kit HR2-112, another five conditions produced glucagon-Cex crystals at 277 K: (i) 10% PEG 6000, 0.1 M HEPES pH 7.5, 5% MPD, (ii) 1.0 M sodium acetate, 0.1 M HEPES pH 7.5, 0.05 M cadmium sulfate, (iii) 10% PEG 8000, 0.1 M HEPES pH 7.5, 8% ethylene glycol, (iv) 20% PEG 10 000, 0.1 M HEPES pH 7.5 and (v) 10% PEG 20 000, 1.0 M Bicine pH 9.0, 2% dioxane (Fig. 2 ▶).
Figure 2.

Crystals of glucagon-Cex.
Based on the above conditions, new sitting-drop vapor-diffusion reservoirs were set up. The sitting drops contained 5 µl protein solution plus 5 µl reservoir solution. The best conditions for final crystallization were (i) 0.1 M succinic acid pH 7.0, 15%(w/v) PEG 3350, (ii) 8% PEG 8000, 0.1 M Tris–HCl pH 8.5 and (iii) 10% PEG 8000, 0.1 M HEPES pH 7.5, 8% ethylene glycol. Using the selenomethionyl peptide, similar conditions always produced crystal showers. To reduce nucleation, the peptide concentration was lowered to 2–5 mg ml−1 solution under the same conditions as described above.
Glucagon-Cex is very soluble, with a high propensity for crystallization. The crystallization is not pH-sensitive and the peptide can be crystallized over a broad pH range from 4.5 to 9.0. PEG 3350, PEG 10 000 and PEG 8000 are the preferred precipitation agents; however, the peptide can also be crystallized from ammonium sulfate alone. This ease of crystallization over a broad pH and PEG molecular-weight range make glucagon-Cex an ideal system for the study of the crystallization mechanism and dynamics of peptide hormones.
2.3. Data collection and preliminary X-ray analysis
For cryogenic data collection, a selenomethionyl glucagon-Cex crystal was flash-frozen in liquid nitrogen in a cryoprotectant solution containing 35% PEG 3350. X-ray data were collected at 100 K at beamline 19BM of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. Two-wavelength inverse-beam SAD data (peak, 0.9790 Å; inflection point, 0.9791 Å) were collected from an SeMet-labeled glucagon-Cex crystal to a Bragg spacing of 2.6 Å; native data were collected to 2.2 Å resolution. The diffraction data were indexed in space group P23, with unit-cell parameters a = b = c = 48.20 Å, α = β = γ = 90.0°. All data were processed and scaled using HKL-2000 to R merge values of 3.0%, 8.9% and 9.1% for the native, inflection point and peak data, respectively (Table 1 ▶). The crystal contains six monomer molecules in the asymmetric unit with a V M (Matthews, 1968 ▶) of 2.16 Å3 Da−1.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| SAD data | |||
|---|---|---|---|
| Peak | Inflection point | Native | |
| Space group | P23 | ||
| Wavelength (Å) | 0.97896 | 0.97910 | 1.000 |
| Unit-cell parameters | |||
| a (Å) | 48.16 | 48.12 | 48.20 |
| b (Å) | 48.16 | 48.12 | 48.20 |
| c (Å) | 48.16 | 48.12 | 48.20 |
| β (°) | 90.0 | 90.0 | 90.0 |
| Resolution range | 50.0–2.6 (2.69–2.60) | 20.0–2.7 (2.80–2.70) | 50.0–2.2 (2.28–2.20) |
| Total reflections | 29780 | 23905 | 35050 |
| Unique reflections | 1275 | 1132 | 2006 |
| I/σ(I) | 21.4 (2.8) | 19.3 (2.54) | 18.6 (2.2) |
| Completeness (%) | 99.4 (100) | 99.7 (99.2) | 99.1 (99.5) |
| Rmerge† | 0.089 (0.410) | 0.091 (0.423) | 0.030 (0.560) |
R
merge = 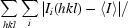
 , where I
i is the intensity of an individual measurement and 〈I〉 is the mean intensity of all measurements of I.
, where I
i is the intensity of an individual measurement and 〈I〉 is the mean intensity of all measurements of I.
3. Discussion
The novel peptide hormone glucagon-Cex was synthesized and successfully crystallized in space group P23 with a diffraction limit of 2.2 Å resolution. A selenomethionyl crystal was used for phasing and structure determination is under way. The space group has some ambiguity at this point. Although the data indexed well in the cubic space group, we cannot rule out the possibility of the lower symmetry space group P21, which gives slightly improved data statistics. The molecular packing and the conformation of the Cex extension will provide some insights into the unique biophysical properties of this peptide. Consequently, the structure will aid in the design of more potent and stable therapeutic glucagon or GLP-1 analogs.
Acknowledgments
We thank Dr Rongguang Zhang and the staff of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory for their assistance during data collection and Dr Jay Nix at MBC/ALS for help during MAD data collection.
References
- Bolli, G. B. (1984). Diabetologia, 27, 485. [DOI] [PubMed] [Google Scholar]
- Doyle, M. E., Theodorakis, M. J., Holloway, H. W., Bernier, M., Greig, N. H. & Egan, J. M. (2003). Regul. Pept.114, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estall, J. L. & Drucker, D. J. (2006). Curr. Pharm. Des.12, 1731–1750. [DOI] [PubMed] [Google Scholar]
- Fanelli, C. G., Porcellati, F., Rossetti, P. & Bolli, G. B. (2006). Nutr. Metab. Cardiovasc. Dis.16, Suppl. 1, S28–S34. [DOI] [PubMed] [Google Scholar]
- Fehse, F., Trautmann, M., Holst, J. J., Halseth, A. E., Nanayakkara, N., Nielsen, L. L., Fineman, M. S., Kim, D. D. & Nauck, M. A. (2005). J. Clin. Endocrinol. Metab.90, 5991–5997. [DOI] [PubMed] [Google Scholar]
- Gearhart, M. M. & Parbhoo, S. K. (2006). AACN Clin. Issues, 17, 50–55. [DOI] [PubMed] [Google Scholar]
- Goke, R., Fehmann, H. C., Linn, T., Schmidt, H., Krause, M., Eng, J. & Goke, B. (1993). J. Biol. Chem.268, 19650–19655. [PubMed] [Google Scholar]
- Jin, L., Briggs, S. L., Chandrasekhar, S., Chirgadze, N. Y., Clawson, D. K., Schevitz, R. W., Smiley, D. L., Tashjian, A. H. & Zhang, F. (2000). J. Biol. Chem.275, 27238–27244. [DOI] [PubMed] [Google Scholar]
- Landsberg, L. (2006). Clin. Exp. Pharmacol. Physiol.33, 863–867. [Google Scholar]
- Larsen, P. J., Vrang, N. & Tang-Christensen, M. (2003). Curr. Pharm. Des.9, 1373–1382. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Meier, J. J., Gallwitz, B., Schmidt, W. E. & Nauck, M. A. (2002). Eur. J. Pharmacol.440, 269–279. [DOI] [PubMed] [Google Scholar]
- Murphy, K. G., Dhillo, W. S. & Bloom, S. R. (2006). Endocr. Rev.27, 719–727. [DOI] [PubMed] [Google Scholar]
- Nauck, M. A. (1998). Acta Diabetol.35, 117–129. [DOI] [PubMed] [Google Scholar]
- Nauck, M. A., Holst, J. J., Willms, B. & Schmiegel, W. (1997). Exp. Clin. Endocrinol. Diabetes, 105, 187–195. [DOI] [PubMed] [Google Scholar]
- Neidigh, J. W., Fesinmeyer, R. M., Prickett, K. S. & Andersen, N. H. (2001). Biochemistry, 40, 13188–13200. [DOI] [PubMed] [Google Scholar]
- Schepp, W., Schmidtler, J., Riedel, T., Dehne, K., Schusdziarra, V., Holst, J. J., Eng, J., Raufman, J. P. & Classen, M. (1994). Eur. J. Pharmacol.269, 183–191. [DOI] [PubMed] [Google Scholar]
- Schnolzer, M., Alewood, P., Jones, A., Alewood, D. & Kent, S. B. (1992). Int. J. Pept. Protein Res.40, 180–193. [DOI] [PubMed] [Google Scholar]
- Wyatt, S. B., Winters, K. P. & Dubbert, P. M. (2006). Am. J. Med. Sci.331, 166–174. [DOI] [PubMed] [Google Scholar]