

The crystallization of HLA-B*1402 in complex with two peptides is reported.
Keywords: HLA-B14 subtypes, major histocompatibility complex polymorphism, HLA-B*1402, subtype-dependent peptide-binding modes, ankylosing spondylitis
Abstract
The product of the human major histocompatibility (HLA) class I allele HLA-B*1402 only differs from that of allele HLA-B*1403 at amino-acid position 156 of the heavy chain (Leu in HLA-B*1402 and Arg in HLA-B*1403). However, both subtypes are known to be differentially associated with the inflammatory rheumatic disease ankylosing spondylitis (AS) in black populations in Cameroon and Togo. HLA-B*1402 is not associated with AS, in contrast to HLA-B*1403, which is associated with this disease in the Togolese population. The products of these alleles can present peptides with Arg at position 2, a feature shared by a small group of other HLA-B antigens, including HLA-B*2705, the prototypical AS-associated subtype. Complexes of HLA-B*1402 with a viral peptide (RRRWRRLTV, termed pLMP2) and a self-peptide (IRAAPPPLF, termed pCatA) were prepared and were crystallized using polyethylene glycol as precipitant. The complexes crystallized in space groups P21 (pLMP2) and P212121 (pCatA) and diffracted synchrotron radiation to 2.55 and 1.86 Å resolution, respectively. Unambiguous solutions for both data sets were obtained by molecular replacement using a peptide-complexed HLA-B*2705 molecule (PDB code 1jge) as a search model.
1. Introduction
Major histocompatibility complex (MHC) class I molecules consist of a highly polymorphic MHC-encoded heavy chain (HC; M r ≃ 32 000) that is noncovalently associated with an invariant protein [β2-microglobulin (β2m), M r ≃ 12 000]. The HC forms a groove that is able to bind peptides, usually with a length of 8–13 amino acids, in an allele-dependent fashion (Madden, 1995 ▶). These peptides are typically derived from fragmented intracellular proteins that may also include viral antigens in the case of a virus-infected cell. The involvement of the MHC in disorders that are characterized by cellular or antibody responses against self-antigens (autoimmune diseases) is insufficiently understood. This is exemplified by the human MHC class I allele HLA-B27 and ankylosing spondylitis (AS; Brewerton et al., 1973 ▶; Schlosstein et al., 1973 ▶), which exhibits the strongest known association of a disease with an HLA class I allele (Horton et al., 2004 ▶). Recently, HLA-B*1403, an allele present only in African populations of Cameroon and Togo, was reported to be associated with AS in the Togolese population (López-Larrea et al., 2002 ▶), where both this disease and HLA-B27 are rare. The closely related allele HLA-B*1402 has a widespread distribution that also includes Caucasians, but has never been found to be associated with AS. The products of these subtypes differ only at residue 156 (Leu in HLA-B*1402, Arg in HLA-B*1403). HLA-B*2705 differs from the two HLA-B14 HCs at 18 amino-acid positions located within the first two domains (α1 and α2) of the extracellular region of the molecules.
A considerable research effort has been devoted towards unravelling the molecular basis of this disease association, but no in-depth understanding of AS pathogenesis has yet been obtained (Khan & Ball, 2002 ▶; Ramos & López de Castro, 2002 ▶; López de Castro, 2007 ▶). As there are suggestions that the recognition of HLA class I complexes by cytotoxic T lymphocytes (CTL) or natural killer cells might play a role in the pathogenesis (Benjamin & Parham, 1990 ▶; Ramos & López de Castro, 2002 ▶; López de Castro, 2007 ▶), peptide presentation by these molecules has provided a research focus in the last decade. AS patients with the HLA-B*2705 subtype have been found to possess elevated levels of CTL directed against the self-peptide pVIPR (RRKWRRWHL, derived from vasoactive intestinal peptide type 1 receptor, residues 400–408), about one sixth of which cross-react with HLA-B*2705-positive cells presenting the viral pLMP2 peptide (RRRWRRLTV, derived from latent membrane protein 2 of Epstein–Barr virus, residues 236–244) (Fiorillo et al., 2000 ▶, 2005 ▶). Individuals with another subtype, HLA-B*2709, the product of which differs from that of HLA-B*2705 at only one position (Asp116 in HLA-B*2705, His116 in HLA-B*2709), do not possess CTL that cross-react with the self-peptide pVIPR (Fiorillo et al., 2000 ▶), suggesting the existence of an HLA-B27 subtype-dependent correlation with AS pathogenesis.
A major feature of HLA-B27 ligands is the presence of arginine at position 2 (pArg2; Jardetzky et al., 1991 ▶; Madden et al., 1992 ▶; Urban et al., 1994 ▶; López de Castro et al., 2004 ▶), which binds tightly into the so-called B pocket of the molecule’s peptide-binding groove. Since this pocket is made up of residues that are polymorphic among HLA class I molecules, few allotypes other than HLA-B27, such as HLA-B14, bind peptides with pArg2 (Merino et al., 2005 ▶). Two peptides were found to be shared by HLA-B*1402, HLA-B*1403, HLA-B*2705 and HLA-B*2709 (Merino et al., 2005 ▶ and unpublished work). One of these is a self-peptide (pCatA, IRAAPPPLF) derived from the signal sequence (residues 2–10) of cathepsin A.
In contrast to the high degree of overlap among the constitutive peptide repertoires of HLA-B27 subtypes (Ramos et al., 2002 ▶), HLA-B*1402 and HLA-B*1403 differ considerably regarding the peptides that are bound within the binding groove of the molecules (Merino et al., 2005 ▶). However, the HLA-B27-bound and HLA-B14-bound peptide repertoires show even less overlap (about 3–5%; Merino et al., 2005 ▶), although both pairs of subtypes share some important peptide-binding properties, such as the preference for pArg2, with HLA-B27 exhibiting a much higher restriction for this motif. It may therefore be envisaged that a thorough structural study of the HLA-B14 antigens and their comparison with the extensive structural findings in the HLA-B*2705/HLA-B*2709 system (Hülsmeyer et al., 2002 ▶, 2004 ▶, 2005 ▶; Hillig et al., 2004 ▶; Fiorillo et al., 2005 ▶; Rückert et al., 2006 ▶) would enhance our understanding of the differential AS association of these four HLA-B subtypes and would help to shed light on the generality of the arthritogenic peptide theory (López de Castro, 2007 ▶).
2. Materials and methods
2.1. Protein preparation
The nonapeptides pLMP2 (RRRWRRLTV) and pCatA (IRAAPPPLF) were chemically synthesized and purified by reverse-phase HPLC (EZBiolab, Westfield, USA). The HLA-B*1402 HC (Merino et al., 2005 ▶) was cloned into the pHN vector. Both HC and β2m were untagged. They were expressed separately and formed inclusion bodies in Escherichia coli that were solubilized with 50%(w/v) urea. The HLA-B14–peptide complexes were prepared by reconstituting the HC (12 mg), β2m (10 mg) and the respective peptide (4 mg) according to a previously described procedure (Garboczi et al., 1992 ▶). Briefly, the reconstitution mixture (400 ml, containing 400 mM arginine–HCl, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione and 100 mM Tris–HCl pH 7.5) was incubated at 277 K for 8–12 weeks, concentrated using Amicon Ultra-15 concentrators and purified by size-exclusion chromatography with Sephadex 75 (Pharmacia). Fractions containing the respective ternary complex were pooled and their composition (HC, β2m) was surveyed by SDS–PAGE. The protein complexes were then concentrated to 15 mg ml−1 using Amicon Ultra-15 in a buffer system containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.01%(w/v) sodium azide and used for crystallization trials.
2.2. Crystallization and data collection
For the HLA-B*1402–pLMP2 complex, a high-throughput robotic crystallization station (Protein Structure Factory, Berlin, Germany; Heinemann et al., 2000 ▶) was used to screen several standard crystallization screen kits from Hampton Research (Aliso Viejo, USA). Sitting-drop vapour-diffusion crystallization trays with 100 µl reservoir solution and 500 nl drops consisting of equal volumes of reservoir and protein solutions were set up and the trays were kept at 291 K for crystal formation. Crystals of HLA-B*1402–pLMP2 that diffracted to 2.74 Å resolution grew in 3 d over a well containing 20%(w/v) polyethylene glycol (PEG) 10 000 and 0.1 M HEPES buffer pH 7.5. Crystallization trials for the HLA-B*1402–pCatA complex were performed in a hanging-drop vapour-diffusion setup at 291 K (1 ml reservoir solution, 2 µl drops consisting of equal volumes of reservoir and protein solutions), but otherwise under conditions identical to those used for crystallizing HLA-B*1402–pLMP2. Crystals diffracting to 2.35 Å were obtained in 5 d in a well containing 24%(w/v) PEG 10 000, 0.1 M HEPES buffer pH 7.5.
Since the initially grown crystals were too small for X-ray analysis, the crystallization conditions for HLA-B*1402–pLMP2 and HLA-B*1402–pCatA were optimized by varying the molecular weight of the PEG and the concentrations of the various constituents in the hanging-drop trays. Drops were streak-seeded with crushed crystals of the same complex from previous experiments using cat whiskers. Thin plate-like hexagonal crystals of HLA-B*1402–pLMP2 with maximum dimensions of about 150 × 100 × 5 µm grew in 20–22%(w/v) PEG 20 000, 0.1 M HEPES buffer pH 7.5 and 24%(w/v) PEG 8000, 0.1 M HEPES buffer pH 7.5, while needle-shaped crystals of HLA-B*1402–pCatA with maximum dimensions of about 300 × 10 × 5 µm grew under similar conditions in 24–27%(w/v) PEG 20 000 with 0.1 M HEPES pH 7.5. Crystals of both complexes were finally transferred to cryoprotectant solution containing the respective reservoir solution and glycerol. The optimum concentrations of glycerol were found to be 20%(w/v) for HLA-B*1402–pLMP2 crystals and 15%(w/v) for HLA-B*1402–pCatA crystals.
X-ray diffraction data sets were collected for both complexes at 100 K on beamline 14.2 at the BESSY II synchrotron facility in Berlin, Germany using a wavelength of 0.91841 Å. The beamline is equipped with a fast-scanning CCD detector from MAR Research (Norderstedt, Germany). The crystals were exposed for 6.75–10 s during data collection. The HLA-B*1402–pLMP2 crystal was flash-annealed (Harp et al., 1998 ▶) by shielding the cryostream for about 5 s. The crystals of the HLA-B*1402–pLMP2 and HLA-B*1402–pCatA complexes belonged to space groups P21 and P212121 and diffracted to 2.55 and 1.85 Å resolution, respectively. The collected data sets were integrated and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). There are two complexes and one complex per crystal asymmetric unit in the HLA-B*1402–pLMP2 and HLA-B*1402–pCatA crystals, respectively. Their Matthews coefficients and solvent contents (Matthews, 1968 ▶) were calculated to be 2.5 Å3 Da−1 and 50.2%, and 2.6 Å3 Da−1 and 52.2%, respectively, based on an approximate M r of ∼45 000 for each of the complexes. Crystallographic data and X-ray data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics of HLA-B*1402–pLMP2 and HLA-B*1402–pCatA.
Values in parentheses are for the highest resolution shell.
| HLA-B*1402–pLMP2 | HLA-B*1402–pCatA | |
|---|---|---|
| Space group | P21 | P212121 |
| Unit-cell parameters (Å) | a = 53.4, b = 79.9, c = 105.4, α = 90, β = 99.5, γ = 90 | a = 50.8, b = 82.1, c = 110.7, α = 90, β = 90, γ = 90 |
| Solvent content (%) | 50.2 | 52.2 |
| Matthews coefficient† (Å3 Da−1) | 2.5 | 2.6 |
| Resolution (Å) | 30.0–2.55 (2.64–2.55) | 50.0–1.86 (1.93–1.86) |
| Unique reflections | 26733 (2177) | 38359 (3023) |
| Completeness (%) | 93.0 (76.8) | 96.8 (78.0) |
| 〈I〉/〈σ(I)〉 | 7.9 (1.4) | 22.5 (3.5) |
| Rsym‡ | 0.137 (0.457) | 0.049 (0.239) |
| Rmerge§ | 0.138 (0.449) | 0.073 (0.247) |
| Rr.i.m.§ | 0.165 (0.585) | 0.082 (0.294) |
| Rp.i.m.§ | 0.089 (0.371) | 0.036 (0.155) |
| Redundancy | 3.0 (1.8) | 4.6 (2.7) |
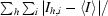
The CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) program Phaser (Storoni et al., 2004 ▶) was used for phase determination of the HLA-B*1402–pLMP2 crystal data employing the atomic coordinates of a high-resolution structure of HLA-B*2705–m9 as a search model (PDB code 1jge; Hülsmeyer et al., 2002 ▶), as MOLREP (Vagin & Teplyakov, 1997 ▶) failed to find a satisfactory solution. HC and β2m coordinates were used as separate ensembles in Phaser and the best final solution obtained by this program was based on top log-likelihood gain (LLG) values of 1609 for HC and 3441 for β2m. The final Z score was over 13 for each of the two HCs and each of the two β2m molecules in the asymmetric unit. The molecular-replacement solution for the HLA-B*1402–pCatA crystal data was obtained by MOLREP using the same high-resolution structure of HLA-B*2705–m9 as a search model (PDB code 1jge; Hülsmeyer et al., 2002 ▶) with the omission of water molecules and peptide.
Models of both HLA-B*1402 complexes pack sensibly in the unit cells and show no clashes. Examination of electron-density maps calculated from the initial phases permitted us to identify the presence of the respective nonapeptides as well as the HC residues unique to HLA-B*1402 in both structures. Modelling of the respective peptides in electron density and rigid-body refinement yielded models with R and R free of 0.262 and 0.342, respectively, and an F o − F c correlation coefficient of 0.882 for HLA-B*1402–pLMP2, and R and R free values of 0.256 and 0.285, respectively, and an F o − F c correlation coefficient of 0.901 for HLA-B*1402–pCatA.
3. Results and discussion
Highly pure HLA-B*1402–pLMP2 and HLA-B*1402–pCatA complexes (Fig. 1 ▶) were crystallized using PEG as precipitant. Crystal formation was optimized by varying the PEG molecular weight and concentration as well as by streak-seeding using crystals of the same complex from previous experiments, which resulted in well ordered crystals (Fig. 2 ▶). X-ray diffraction analysis (Fig. 3 ▶) revealed that the crystals of the two complexes were non-isomorphous and belonged to space groups P21 and P212121, respectively, which are also typical for HLA-B27–peptide complexes (see, for example, Hülsmeyer et al., 2002 ▶, 2004 ▶, 2005 ▶). Previously described crystals of HLA-B27 subtypes with the pLMP2 peptide belong to either space group P21 (HLA-B*2705 and HLA-B*2709; Fiorillo et al., 2005 ▶) or P212121 (HLA-B*2703, HLA-B*2704 and HLA-B*2706; Loll, Zawacka, Biesiadka, Petter et al., 2005 ▶; Loll, Zawacka, Biesiadka, Rückert et al., 2005 ▶; Zawacka et al., 2005 ▶). However, two complexes per asymmetric unit as found for HLA-B*1402–pLMP2 have not been observed in any crystal of HLA-B27–pLMP2. Information with regard to the conformation of the bound peptide cannot be inferred from these similarities or differences, since crystals of HLA-B*2705–pLMP2 and HLA-B*2709–pLMP2, which are near-isomorphous and share the same space group (P21), nevertheless display the pLMP2 peptide in completely different binding modes (Fiorillo et al., 2005 ▶). Modelling of solvent molecules in unassigned electron density is in progress for both structures and complete structural details will be reported separately.
Figure 1.

SDS–PAGE analysis of refolded HLA-B*1402–pLMP2 (lanes 2 and 3, about 75 µg each) and HLA-B*1402–pCatA (lanes 1 and 4, about 20 µg each) complexes. Purified complexes were electrophoretically analyzed under reducing (lanes 1 and 2) and nonreducing conditions (lanes 3 and 4) using Coomassie Brilliant Blue stain. Approximate molecular weights of marker proteins are indicated in kDa on the left. HLA-B*1402 heavy-chain (HC) and β2-microglobulin (β2m) bands are indicated. A faint β2m dimer (d) band is visible under nonreducing conditions (lanes 3 and 4) and even under reducing conditions when very high amounts of HLA–peptide complex are loaded onto a gel (lane 2). This has so far not been observed to hinder crystallization of the complex.
Figure 2.

Crystals of (a) HLA-B*1402–pLMP2 and (b) HLA-B*1402–pCatA complexes exhibit remarkably different morphology and size.
Figure 3.

Diffraction pattern of an HLA-B*1402–pCatA crystal collected at BESSY II synchrotron facility in Berlin.
The structure of the HLA-B*1402 complexes will permit comparison with other previously determined structures of HLA-B27 molecules in complex with these peptides, enabling a direct assessment of the binding mode of the same peptide (pLMP2) in complex with significantly distinct HLA class I molecules. A detailed structural study of peptide presentation by these complexes will improve our understanding of HLA-B subtype-dependent T-cell recognition and might also shed light on the causes underlying the association of certain HLA-B27 and HLA-B14 alleles with spondyloarthropathies.
Acknowledgments
Financial support for this work was provided by Deutsche Forschungsgemeinschaft (SFB 449/B6 and Z3), the European Union (EFRE 2000-2006 2ü/2), Fonds der Chemischen Industrie, Sonnenfeld Stiftung, Berlin, Senate of Berlin (NaFöG fellowship, PK), grant SAF2005-03188 from the Spanish Ministry of Education and Science (EM and JALC) and an institutional grant of the Fundación Ramon Areces to the Centro de Biología Molecular Severo Ochoa. We thank Christina Schnick for excellent technical assistance and are grateful for allocation of beam time and support at BESSY II (Berlin).
References
- Benjamin, R. & Parham, P. (1990). Immunol. Today, 11, 137–142. [DOI] [PubMed] [Google Scholar]
- Brewerton, D. A., Hart, F. D., Nicholls, A., Caffrey, M., James, D. C. & Sturrock, R. D. (1973). Lancet, 1, 904–907. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Fiorillo, M. T., Maragno, M., Butler, R., Dupuis, M. L. & Sorrentino, R. (2000). J. Clin. Invest.106, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo, M. T., Rückert, C., Hülsmeyer, M., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2005). J. Biol. Chem.280, 2962–2971. [DOI] [PubMed] [Google Scholar]
- Garboczi, D. N., Hung, D. T. & Wiley, D. C. (1992). Proc. Natl Acad. Sci. USA, 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harp, J. M., Timm, D. E. & Bunick, G. (1998). Acta Cryst. D54, 622–628. [DOI] [PubMed] [Google Scholar]
- Heinemann, U., Frevert, J., Hofmann, K., Illing, G., Maurer, C., Oschkinat, H. & Saenger, W. (2000). Prog. Biophys. Mol. Biol.73, 347–362. [DOI] [PubMed] [Google Scholar]
- Hillig, R. C., Hulsmeyer, M., Saenger, W., Welfle, K., Misselwitz, R., Welfle, H., Kozerski, C., Volz, A., Uchanska-Ziegler, B. & Ziegler, A. (2004). J. Biol. Chem.279, 652–663. [DOI] [PubMed] [Google Scholar]
- Horton, R., Wilming, L., Rand, V., Lovering, R. C., Bruford, E. A., Khodiyar, V. K., Lush, M. J., Povey, S., Talbot, C. C. Jr, Wright, M. W., Wain, H. M., Trowsdale, J., Ziegler, A. & Beck, S. (2004). Nature Rev. Genet.5, 889–899. [DOI] [PubMed]
- Hülsmeyer, M., Fiorillo, M. T., Bettosini, F., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2004). J. Exp. Med.199, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hülsmeyer, M., Hillig, R. C., Volz, A., Rühl, M., Schröder, W., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2002). J. Biol. Chem.277, 47844–47853. [DOI] [PubMed] [Google Scholar]
- Hülsmeyer, M., Welfle, K., Pöhlmann, T., Misselwitz, R., Alexiev, U., Welfle, H., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). J. Mol. Biol.346, 1367–1379. [DOI] [PubMed] [Google Scholar]
- Jardetzky, T. S., Lane, W. S., Robinson, R. A., Madden, D. R. & Wiley, D. C. (1991). Nature (London), 353, 326–329. [DOI] [PubMed] [Google Scholar]
- Khan, M. A. & Ball, E. J. (2002). Best Pract. Res. Clin. Rheumatol.16, 675–690. [PubMed] [Google Scholar]
- Loll, B., Zawacka, A., Biesiadka, J., Petter, C., Rückert, C., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). Acta Cryst. F61, 939–941. [DOI] [PMC free article] [PubMed]
- Loll, B., Zawacka, A., Biesiadka, J., Rückert, C., Volz, A., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). Acta Cryst. F61, 372–374. [DOI] [PMC free article] [PubMed]
- López de Castro, J. A. (2007). Immunol. Lett.108, 27–33. [DOI] [PubMed] [Google Scholar]
- López de Castro, J. A., Alvarez, I., Marcilla, M., Paradela, A., Ramos, M., Sesma, L. & Vazquez, M. (2004). Tissue Antigens, 63, 424–445. [DOI] [PubMed] [Google Scholar]
- López-Larrea, C., Mijiyawa, M., Gonzalez, S., Fernandez-Morera, J. L., Blanco-Gelaz, M. A., Martinez-Borra, J. & López-Vazquez, A. (2002). Arthritis Rheum.46, 2968–2971. [DOI] [PubMed] [Google Scholar]
- Madden, D. R. (1995). Annu. Rev. Immunol.13, 587–622. [DOI] [PubMed] [Google Scholar]
- Madden, D. R., Gorga, J. C., Strominger, J. L. & Wiley, D. C. (1992). Cell, 70, 1035–1048. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Merino, E., Montserrat, V., Paradela, A. & López de Castro, J. A. (2005). J. Biol. Chem.280, 35868–35880. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ramos, M. & López de Castro, J. A. (2002). Tissue Antigens, 60, 191–205. [DOI] [PubMed] [Google Scholar]
- Ramos, M., Paradela, A., Vazquez, M., Marina, A., Vazquez, J. & López de Castro, J. A. (2002). J. Biol. Chem.277, 28749–28756. [DOI] [PubMed] [Google Scholar]
- Rückert, C., Loll, B., Fiorillo, M. T., Moretti, R., Biesiadka, J., Saenger, W., Ziegler, A., Sorrentino, R. & Uchanska-Ziegler, B. (2006). J. Biol. Chem.281, 2306–2316. [DOI] [PubMed] [Google Scholar]
- Schlosstein, L., Terasaki, P. I., Bluestone, R. & Pearson, C. M. (1973). N. Engl. J. Med.288, 704–706. [DOI] [PubMed] [Google Scholar]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed] [Google Scholar]
- Urban, R. G., Chicz, R. M., Lane, W. S., Strominger, J. L., Rehm, A., Kenter, M. J., UytdeHaag, F. G., Ploegh, H., Uchanska-Ziegler, B. & Ziegler, A. (1994). Proc. Natl Acad. Sci. USA, 91, 1534–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135. [Google Scholar]
- Zawacka, A., Loll, B., Biesiadka, J., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). Acta Cryst. F61, 1097–1099. [DOI] [PMC free article] [PubMed]