

E. coli WrbA, the founding member of a novel flavoprotein family, was crystallized in complex with its physiological cofactor. Preliminary diffraction analysis is reported.
Keywords: flavoproteins, flavin cofactor
Abstract
The flavoprotein WrbA from Escherichia coli is considered to be the prototype of a new family of multimeric flavodoxin-like proteins that are implicated in cell protection against oxidative stress. The present study is aimed at structural characterization of the E. coli protein with respect to its recently revealed oxidoreductase activity. Crystals of WrbA holoprotein in complex with the oxidized flavin cofactor (FMN) were obtained using standard vapour-diffusion techniques. Deep yellow tetragonal crystals obtained from differing crystallization conditions display different space groups and unit-cell parameters. X-ray crystal structures of the WrbA holoprotein have been determined to resolutions of 2.0 and 2.6 Å.
1. Introduction
The protein WrbA from Escherichia coli is the founding member of a family of flavodoxin-like proteins that are suggested by homology to participate in cellular defence against oxidative stress (Grandori & Carey, 1994 ▶). The designation of the E. coli WrbA protein as tryptophan-repressor binding protein A refers to its discovery: it was copurified with the tryptophan-repressor protein TrpR and reported to enhance the stability of DNA binding by TrpR (Yang et al., 1993 ▶). However, this role was put under question by the finding that WrbA exerted no specific effect on TrpR–DNA binding (Grandori et al., 1998 ▶). This work also showed that WrbA binds flavin mononucleotide (FMN) more weakly than the flavodoxins, but that it nevertheless does so specifically, and that it undergoes dimer–tetramer equilibrium in solution. Sequence similarity with NAD(P)H:quinone oxidoreductases suggested that the WrbA family might share this function (Laskowski et al., 2002 ▶; Daher et al., 2005 ▶). Recently, Patridge & Ferry (2006 ▶) reported that E. coli WrbA and its homologue from Archaeoglobus fulgidus display quinone oxidoreductase activity, with FMN as the redox-active cofactor and NADH as the preferred electron donor. Enzymes with NAD(P)H:quinone reductase activity are proposed to maintain quinones in a fully reduced state in order to protect cells against deleterious reactive oxygen species from one-electron redox cycling (Jensen et al., 2002 ▶; Cohen et al., 2004 ▶; Jaiswal, 2000 ▶; Morre, 2004 ▶; Wang et al., 2006 ▶; Gonzalez et al., 2005 ▶; Talalay & Dinkova-Kostova, 2004 ▶; Bianchet et al., 2004 ▶; Ross & Siegel, 2004 ▶). Consistent with this role, E. coli WrbA transfers two electrons at a time and does not stabilize a flavin semiquinone intermediate (Nöll et al., 2006 ▶). Transcription of E. coli WrbA is controlled by the stress response factor rpoS (Lacour & Landini, 2004 ▶). A wide range of external stressors such as acids or H2O2 have been reported to induce WrbA expression (Chang et al., 2002 ▶; Tucker et al., 2002 ▶; Kang et al., 2005 ▶). These results implicate the WrbA family in the protection of cells against oxidative stress.
The crystal structures of WrbA homologues from Deinococcus radiodurans (37% sequence identity with E. coli WrbA) and Pseudomonas aeruginosa (40% sequence identity with E. coli WrbA) have been published (Gorman & Shapiro, 2005 ▶). These structures confirm that the members of the WrbA family adopt an α/β twisted open-sheet fold typical of flavodoxins and form homotetramers that bind one FMN molecule per monomer. However, the structures did not clarify why WrbA proteins function as tetramers or why they bind FMN specifically but only weakly. Furthermore, the only high-resolution structure to be reported is that of the D. radiodurans apoprotein; the holoprotein form is of much lower resolution and both the forms of the P. aeruginosa protein are poorly ordered, with many missing chain segments. D. radiodurans is a eubacterial extremophile and its WrbA structure may reflect adaptations of sequence and structure required for radiation stability. This fact, together with its relatively low sequence identity to E. coli WrbA, limits the structural interpretation of the biochemical data, which are available only for the E. coli WrbA protein. For these reasons, we have been pursuing the crystallization of E. coli WrbA. We have previously reported the crystallization of E. coli WrbA apoprotein (Wolfova et al., 2005 ▶). Here, we report the crystallization and preliminary diffraction analysis of E. coli WrbA in complex with its FMN cofactor.
2. Materials and methods
2.1. Expression, purification and holoprotein reconstitution
Recombinant WrbA protein was expressed in E. coli CY15071(λDE3) cells and purified using DEAE-cellulose (DEAE Highprep, Pharmacia) and Affi-Gel Blue (Bio-Rad) affinity column chromatography, as previously described by Grandori et al. (1998 ▶) and Natalello et al. (2007 ▶). The purity and monomer molecular weight (about 21 kDa) were determined by electrospray-ionization mass spectrometry (ESI–MS) and by SDS–PAGE performed according to Laemmli (1970 ▶) in a Bio-Rad minigel apparatus using 12% Tris–glycine gels stained with Coomassie Blue R250. The final concentration of the purified protein was determined by UV absorption spectroscopy (Spectronic Unicam UV 300, Cambridge, England) using the previously reported extinction coefficient of 22 831 M −1 cm−1 at 280 nm (Grandori et al., 1998 ▶). Pure WrbA protein was obtained without bound cofactor owing to loss of FMN during affinity chromatography. The holoprotein for crystallization was prepared by incubating 0.25 mM pure WrbA apoprotein with 0.25 mM FMN (Sigma), affording 96% occupancy of the FMN–WrbA complex according to the equilibrium constant of 2 µM as determined by Grandori et al. (1998 ▶). The use of an excess of cofactor over WrbA protein for reconstitution was prohibited owing to precipitation of free FMN, which prevented the protein from crystallizing (data not shown). The reconstituted WrbA holoprotein is in the oxidized form as shown by the absorption spectra (Grandori et al., 1998 ▶), which are typical of oxidized flavoproteins (Mayhew & Massey, 1969 ▶; Mayhew & Tollin, 1992 ▶).
2.2. Crystallization
Reconstituted WrbA holoprotein at 0.25 mM (5 mg ml−1) in 20 mM Tris–HCl pH 7.5 was used for the crystallization trials. As a starting point, the crystallization conditions found previously for WrbA apoprotein were tested (reservoir containing 2.0 M ammonium sulfate in 0.1 M Tris–HCl pH 8.5; Wolfova et al., 2005 ▶). Screening of varying temperatures (ranging from 277 to 298 K) and concentrations of ammonium sulfate (1.5–2.6 M at pH values ranging from 7.0 to 9.0) was carried out using the sitting-drop vapour-diffusion technique. Crystallization trials using droplets containing equal volumes of protein and precipitant solution were performed in Cryschem plates (Hampton Research, Aliso Viejo, CA, USA). In addition, 0.5–2.0 mM cadmium chloride and 1.0 mM lithium citrate were tested as additives because of their positive effect on WrbA apoprotein crystallization (Wolfova et al., 2005 ▶). Commercially available crystal screening kits (Hampton Research Crystal Screen Kit, Sigma Basic and Extension Crystallization Kits for Proteins) were also used for crystallization trials; screening was carried out using 96-well Intelliplates and the Phoenix microdispenser (Art Robbins Instruments).
2.3. Data collection and processing
Diffraction data for WrbA holoprotein crystals were collected using synchrotron radiation at the Joint University of Hamburg/IMB Jena/EMBL beamline X13 (wavelength 0.804 Å), Deutsches Elektronen-Synchrotron (DESY, Hamburg), which is equipped with a MAR CCD detector (X-ray Research). Crystals grown from the conditions reported here were mounted directly into cryoloops without additional cryoprotection and flash-frozen in a stream of nitrogen gas (Oxford Cryosystems) at 100 K. The addition of cryoprotectant was not required since the crystals were obtained at high polyethylene glycol concentrations. X-ray data were recorded to a Bragg spacing of 2.6 Å for crystals grown from 25% ethylene glycol (crystal form I) and to 2.0 Å for crystals grown from 20% PEG 8000 in 0.1 M Tris–HCl pH 8.0 or 30% PEG 4000 in 0.1 M Tris–HCl pH 8.5, 0.2 M MgCl2 (crystal form II). The programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶) were used for processing, indexing and scaling of the data.
2.4. Preliminary structure solution
The structure of crystal form I (space group P43212, diffracting to 2.6 Å; Table 1 ▶) was solved by molecular-replacement techniques using P. aeruginosa WrbA holoprotein as the template (PDB ID code 1zwl; Gorman & Shapiro, 2005 ▶). The structure of crystal form II (space group P41212, diffracting to 2.0 Å; Table 1 ▶) was solved using a partially refined structure of the first crystal form as a search model for molecular replacement. Self- and cross-rotation functions, translational searches and rigid-body refinement were performed using the programs AMoRe (Navaza, 2001 ▶) and MOLREP (Vagin & Teplyakov, 2000 ▶) as implemented in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
Table 1. Data-collection statistics for WrbA holoprotein crystals.
Values in parentheses are for the highest resolution shell.
| WrbA–FMN† | WrbA–FMN‡ | |
|---|---|---|
| Space group | P43212 | P41212 |
| Crystal system | Primitive tetragonal | Primitive tetragonal |
| Unit-cell parameters (Å) | a = b = 94.35, c = 175.38 | a = b = 61.13, c = 168.38 |
| Resolution range (Å) | 35.0–2.6 (2.69–2.60) | 35.0–2.0 (2.04–1.99) |
| Solvent content (%) | 73 | 34 |
| No. of collected images | 180 | 270 |
| Oscillation angle (°) | 1 | 1 |
| No. of measured reflections | 179066 | 142741 |
| No. of unique reflections | 24986 | 20763 |
| Redundancy | 7.2 (6.7) | 6.9 (6.5) |
| Completeness (%) | 98.9 (98.1) | 90.8 (84.1) |
| Rmerge§ | 0.14 (0.62) | 0.09 (0.51) |
| 〈I/σ(I)〉 | 12.35 (2.64) | 16.94 (3.42) |
| Mosaicity (°) | 0.38 | 0.84 |
| No. of molecules per ASU | 2 | 2 |
WrbA holoprotein crystal form I grown from 25% ethylene glycol.
WrbA holoprotein crystal form II grown from 20% PEG 8000, 0.1 M Tris–HCl pH 8.0. Similar data were obtained for crystals grown from 30% PEG 4000, 0.1 M Tris–HCl pH 8.5, 0.2 M MgCl2 (data not shown).
R
merge = 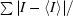
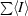 , where I is an individual intensity measurement and 〈I〉 is the average intensity for this reflection. The value of R
merge was calculated by summation over all data.
, where I is an individual intensity measurement and 〈I〉 is the average intensity for this reflection. The value of R
merge was calculated by summation over all data.
3. Results and discussion
3.1. Crystallization and diffraction data
Crystallization conditions based on those found for WrbA apoprotein (Wolfova et al., 2005 ▶) were tested for WrbA holoprotein. However, these conditions yielded no crystals of WrbA holoprotein. Therefore, further screening was carried out using standard crystallization screening kits in 96-well sitting-drop vapour-diffusion experiments at different temperatures. After two weeks, well formed tetragonal crystals grew at 285 K from three different conditions (25% ethylene glycol; 20% PEG 8000, 0.1 M Tris–HCl pH 8.0; 30% PEG 4000, 0.1 M Tris–HCl pH 8.5, 0.2 M MgCl2). The crystals reached maximum dimensions of 0.48 × 0.2 × 0.2 mm and were deep yellow in colour, indicating that the FMN cofactor is present in its oxidized form (Fig. 1 ▶). Thus, the flavin cofactor in the crystals retained the redox state observed by absorption spectroscopy after holoprotein reconstitution (Grandori et al., 1998 ▶). The holoprotein crystals grown under the conditions reported here required no additional optimization steps to yield high-quality diffraction data. In contrast, crystallization of WrbA apoprotein required the use of additives and gels to reduce formation of multicrystalline clusters (Wolfova et al., 2005 ▶). Although optimization yielded single crystals, the apoprotein structure could not be solved owing to high crystal mosaicity and anisotropy.
Figure 1.

WrbA holoprotein crystals grown by the sitting-drop vapour-diffusion method from reservoirs containing: (a) 25% ethylene glycol (crystal form I), (b) 20% PEG 8000, 0.1 M Tris–HCl pH 8.0 (crystal form II), (c) 30% PEG 4000, 0.1 M Tris–HCl pH 8.5, 0.2 M MgCl2.
Diffraction data collected from holoprotein crystals obtained under different crystallization conditions revealed two forms of primitive tetragonal crystals (see §3.2) that differed in space group and unit-cell parameters. The data-collection statistics are presented in Table 1 ▶. Crystals grown from the two conditions containing PEG provided similar diffraction data; hence, only data for the crystals grown from 20% PEG 8000, 0.1 M Tris–HCl pH 8.0 are presented. The crystals grown from 25% ethylene glycol belong to space group P43212, with unit-cell parameters a = b = 94.35, c = 175.38 Å. The crystals grown from 20% PEG 8000 belong to space group P41212, with unit-cell parameters a = b = 61.13, c = 168.38 Å. The resolution of the diffraction data obtainable with these two crystal forms is also rather different: 2.6 and 2.0 Å, respectively. The very different unit cells of the two crystal forms yield correspondingly large differences in the crystal volume per unit of molecular weight, V M, which is 4.6 and 1.85 Å3 Da−1, respectively. These values correspond to solvent contents of 73% and 34% (Matthews, 1968 ▶). The low protein content explains the lower resolution of the data obtained from the crystals grown from 25% ethylene glycol. The diffraction limit of the crystals grown from PEG 8000 represents the best resolution reported to date for WrbA holoprotein.
3.2. Structure solution of the WrbA–FMN complexes
3.2.1. Structure solution of crystal form I grown from 25% ethylene glycol
Inspection of axial reflections indicates one fourfold screw, one twofold screw and one twofold axis, suggesting either space group P41212 or P43212. Fig. 2 ▶ shows the stereographic projection of the κ = 180° section of the self-rotation function for this crystal form. The crystallographic P422 symmetry corresponds to the peak at the centre and peaks at the edge of this projection. The noncrystallographic twofold symmetry follows from the four equivalent peaks around the central peak on this section. The correct space group P43212 was deduced from a comparison of solutions from the translation function for the enantiomorphic space groups. Using a monomer of P. aeruginosa holoprotein (PDB code 1zwl; monomer plus bound FMN but without water molecules) as the search model in molecular replacement, a dimer was identified consisting of the two best monomer solutions obtained (dimer solution, R = 48.6%, CC = 44.2%; first incorrect solution, R = 52.3%, CC = 28.6%). After rigid-body refinement (REFMAC5; Murshudov et al., 1997 ▶), the initial R factor of 47.8% fell to 39.7% (R free = 42.7%) in the very first cycles of refinement and rebuilding. The asymmetric part of the unit cell contains two chemically identical molecules that form a tetramer with a symmetry-related dimer. The tetramer assembles as a dimer of dimers of 222 symmetry with the twofold axes (noncrystallographic) oriented diagonally to the crystallographic axes; this is also nicely visible from the stereographic projection of the self-rotation function (Fig. 2 ▶).
Figure 2.

Stereographic projection of the κ = 180° section of the self-rotation function calculated using data for P43212 crystals (crystal form I) in the range 40.0–4.0 Å and an integration radius of 20 Å.
3.2.2. Structure solution of crystal form II grown from 20% PEG 8000
Inspection of axial reflections implies one fourfold screw, one twofold screw and one twofold axis. The correct space group P41212 was deduced from a comparison of solutions from the translation function for the enantiomorphic space groups (P41212/P43212 with CC = 62.5%/34.3%). In this case, the structure of crystal form I was used as a search model in molecular replacement. Again, a dimer was identified consisting of the two best monomer solutions (dimer solution, R = 40.2%, CC = 62.5%; first incorrect solution, R = 40.5%, CC = 61.8%). This solution was further improved by rigid-body refinement (MOLREP; R = 38.7%, CC = 65.0%). This dimer also forms a tetramer, i.e. a dimer of dimers with 222 symmetry. However, the noncrystallographic twofold axes are oriented differently compared with crystal form I.
3.3. Preliminary analysis of crystal packing
Preliminary analysis of the diffraction data provided an approximate view of the structural models. The asymmetric unit of both crystal forms contains a dimer that forms a compact tetramer with two adjacent monomers, which is consistent with evidence that E. coli WrbA undergoes a dimer–tetramer equilibrium in solution (Grandori et al., 1998 ▶). Fourier-transform infrared spectroscopy suggests a reduction of conformational heterogeneity and/or dynamics of WrbA upon FMN binding and mass spectroscopy reveals that FMN binding favours tetramer formation from WrbA dimers while not affecting the monomer–dimer distribution (Natalello et al., 2007 ▶). FMN is also proposed to favour tetramer formation by WrbA homologues (Gorman & Shapiro, 2005 ▶).
Ligand binding is among various factors that affect protein crystallization, as summarized by Ducruix & Giegé (1999 ▶). Here, FMN is suggested to influence the crystallization of WrbA through specific binding leading to favourable intermolecular interactions and/or by reducing the conformational heterogeneity of the protein originating from dynamics of secondary, tertiary or quaternary structure or from aggregation. For the structures reported here, the bound FMN cofactor appears to affect a nearby surface-exposed mobile helix and loop involved in crystal lattice formation (see Fig. 3 ▶). Through specific interactions with WrbA, FMN apparently restricts the conformational degrees of freedom of this part of the chain so that chain excursions and/or species distributions are reduced. Notably, FMN protects WrbA against deliberate proteolysis, resulting in a longer persistence of the holoprotein than the apoprotein (Wolfova & Carey, unpublished observations). Thus, the holoprotein crystals reported here differ fundamentally from those used in the previously reported structures, in which FMN was soaked into crystals of apoprotein (Gorman & Shapiro, 2005 ▶).
Figure 3.

Crystal packing of the WrbA holoprotein in crystal form I grown from 25% ethylene glycol. Each dimer is uniquely coloured. FMN is shown in space-filling representation and the surface-exposed helix discussed in the text is indicated.
The entire polypeptide chain appears to be resolved in the 2.6 Å resolution structure. Thus, for the first time all loops are fully visible in the electron-density maps. In contrast, a few disordered parts are present in the high-resolution model. Consequently, in order to obtain maximum structural information, both structural models will have to be pursued. The two structures are currently in the process of being refined and interpreted in detail.
Acknowledgments
The authors thank Rolf Hilgenfeld (University of Luebeck) and Juraj Sedláček (Academy of Sciences of the Czech Republic, Prague) for their generous support, and Rüdiger Ettrich for his advice and comments. This work is supported by the Ministry of Education of the Czech Republic (projects Kontakt ME640, MSM6007665808 and LC06010), by the Academy of Sciences of the Czech Republic (AV0Z60870520) and by NSF INT 03-09049 to JC. Diffraction measurements at the Deutsches Elektronen-Synchrotron (DESY, Hamburg) were supported by the European Community, Research Infrastructure Action under the FP6 ‘Structuring the European Research Area Specific Programme’ to the EMBL Hamburg Outstation, contract No. RII3-CT-2004-506008.
References
- Bianchet, M. A., Faig, M. & Amzel, L. M. (2004). Methods Enzymol.382, 144–174. [DOI] [PubMed] [Google Scholar]
- Chang, D. E., Smalley, D. J. & Conway, T. (2002). Mol. Microbiol.45, 289–306. [DOI] [PubMed] [Google Scholar]
- Cohen, R., Suzuki, M. R. & Hammel, K. E. (2004). Appl. Environ. Microbiol.70, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Daher, B. S., Venancio, E. J., de Freitas, S. M., Bao, S. N., Vianney, P. V., Andrade, R. V., Dantas, A. S., Soares, C. M., Silva-Pereira, I. & Felipe, M. S. (2005). Fungal Genet. Biol.42, 434–443. [DOI] [PubMed] [Google Scholar]
- Ducruix, A. & Giegé, R. (1999). Crystallization of Nucleic Acids and Proteins: A Practical Approach, 2nd ed. Oxford University Press.
- Gonzalez, C. F., Ackerley, D. F., Lynch, S. V. & Matin, A. (2005). J. Biol. Chem.280, 22590–22595. [DOI] [PubMed] [Google Scholar]
- Gorman, J. & Shapiro, L. (2005). Protein Sci.14, 3004–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori, R. & Carey, J. (1994). Protein Sci.3, 2185–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori, R., Khalifah, P., Boice, J. A., Fairman, R., Giovanelli, K. & Carey, J. (1998). J. Biol. Chem.273, 20960–20966. [DOI] [PubMed] [Google Scholar]
- Jaiswal, A. K. (2000). Free Radic. Biol. Med.29, 254–262. [DOI] [PubMed] [Google Scholar]
- Jensen, K. A. Jr, Ryan, Z. C., Vanden Wymelenberg, A., Cullen, D. & Hammel, K. E. (2002). Appl. Environ. Microbiol.68, 2699–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y., Weber, K. D., Qiu, Y., Kiley, P. J. & Blattner, F. R. (2005). J. Bacteriol.187, 1135–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour, S. & Landini, P. (2004). J. Bacteriol.186, 7186–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Laskowski, M. J., Dreher, K. A., Gehring, M. A., Abel, S., Gensler, A. L. & Sussex, I. M. (2002). Plant Physiol.128, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Mayhew, S. G. & Massey, V. (1969). J. Biol. Chem.244, 794–802. [PubMed] [Google Scholar]
- Mayhew, S. G. & Tollin, G. (1992). Chemistry and Biochemistry of Flavoenzymes, Vol. 3, edited by F. Mueller, pp. 389–426. Boca Raton: CRC Press.
- Morre, D. J. (2004). Methods Enzymol.378, 179–199. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Natalello, A., Doglia, S. M., Carey, J. & Grandori, R. (2007). Biochemistry, 46, 543–553. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Nöll, G., Kozma, E., Grandori, R., Carey, J., Schödl, T., Hauska, G. & Daub, J. (2006). Langmuir, 22, 2378–2383. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Patridge, E. V. & Ferry, J. G. (2006). J. Bacteriol.188, 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, D. & Siegel, D. (2004). Methods Enzymol.382, 115–144. [DOI] [PubMed] [Google Scholar]
- Talalay, P. & Dinkova-Kostova, A. T. (2004). Methods Enzymol.382, 355–364. [DOI] [PubMed] [Google Scholar]
- Tucker, D. L., Tucker, N. & Conway, T. (2002). J. Bacteriol.184, 6551–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Wang, G., Alamuri, P. & Maier, R. J. (2006). Mol. Microbiol.61, 847–860. [DOI] [PubMed] [Google Scholar]
- Wolfova, J., Grandori, R., Kozma, E., Chatterjee, N., Carey, J. & Kuta Smatanova, I. (2005). J. Cryst. Growth, 284, 502–505.
- Yang, W., Ni, L. & Somerville, R. L. (1993). Proc. Natl Acad. Sci. USA, 90, 5796–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]