

The ligand-binding domain of metabotropic glutamate receptor 7 has been overexpressed, purified, and crystallized by the hanging-drop vapour-diffusion method. A complete data set has been collected to 3.30 Å.
Keywords: metabotropic glutamate receptors, ligand-binding domains
Abstract
Glutamate is the major excitatory neurotransmitter and its metabotropic glutamate receptor (mGluR) plays an important role in the central nervous system. The ligand-binding domain (LBD) of mGluR subtype 7 (mGluR7) was produced using the baculovirus expression system and purified from the culture medium. The purified protein was characterized by gel-filtration chromatography, SDS–PAGE and a ligand-binding assay. Crystals of mGluR7 LBD were grown at 293 K by the hanging-drop vapour-diffusion method. The crystals diffracted X-rays to 3.30 Å resolution using synchrotron radiation and belong to the trigonal space group P3121, with unit-cell parameters a = b = 92.4, c = 114.3 Å. Assuming the presence of one protomer per crystallographic asymmetric unit, the Matthews coefficient V M was calculated to be 2.5 Å3 Da−1 and the solvent content was 51%.
1. Introduction
Metabotropic glutamate receptors (mGluRs) are involved in the regulation of many physiological and pathological processes in the central nervous system, including synaptic plasticity, learning and memory, pain transmission and neurodegeneration. The mGluRs are principal members of the class C G-protein-coupled receptors. The eight mGluR subtypes are divided into three subgroups (group I, II, and III) according to sequence homology, associated second messenger systems and pharmacological properties (Nakanishi & Masu, 1994 ▶; Hollmann & Heinemann, 1994 ▶).
The mGluRs consist of three domains: the N-terminal extracellular, seven transmembrane and the C-terminal cytoplasmic domains. The N-terminal extracellular domain contains ligand-binding and cysteine-rich regions and forms a homodimer. Crystal structures of the extracellular ligand-binding domain (LBD) of the homodimeric mGluR1 (group I) with or without agonist/antagonist have been reported previously (Kunishima et al., 2000 ▶; Tsuchiya et al., 2002 ▶). Both ‘active’ and ‘resting’ conformations were found in the ligand-free state, suggesting that an equilibrium between these two conformations exists in the absence of ligands. The structures with ligands revealed that the ‘active’ form is induced by glutamate binding and that the antagonist stabilizes the ‘resting’ conformation. In order to validate the application of this proposal to all mGluRs, three-dimensional structures from other subgroups are required. Moreover, the structure of an mGluR from another group may clarify the discrimination of the group-specific ligands in terms of three-dimensional structures and this information may serve in the development of new drugs.
Here, we report the expression, purification, crystallization and preliminary X-ray crystallographic analysis of mGluR7 LBD (group III). We expressed mGluR7 LBD in insect cells using the baculovirus expression system and purified it from the culture supernatant. The oligomeric state and ligand-binding ability of the purified protein were characterized. We were also successful in obtaining crystals suitable for structure determination at atomic resolution.
2. Materials and methods
2.1. Expression and purification of the ligand-binding domain of mGluR7
The mGluR7 LBD encompassing amino-acid residues Met1–Ser521 from Rattus norvegicus (PIR accession No. P35400; Okamoto et al., 1994 ▶) was cloned by the PCR method using full-length mGluR7 cDNA as a template. At the same time, an oligonucleotide sequence coding for the amino-acid residues LVPRGSHHHHHH was introduced into the PCR primer for the attachment of a His-tag next to the C-terminus of mGluR7 LBD. In addition, the N-terminal signal sequence was expected to be removed during the secretion process. Recombinant baculovirus was constructed using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer’s protocol. In order to produce the target protein, Trichoplusia ni BTI-TN-5B1-4 (High Five, Invitrogen) cells were cultured in a monolayer using Express Five serum-free medium (Invitrogen) supplemented with 16.5 mM l-glutamine. Baculovirus infection was performed as described previously (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶). The culture medium into which the target protein was successfully secreted was collected 4–5 d after inoculation.
After the addition of protease inhibitors (5 µM E-64 and 1 mM phenylmethylsulfonyl fluoride), the medium containing the High Five cells was filtered using a 0.22 µm membrane and loaded onto Ni–NTA agarose (Qiagen) packed in an XK26 column (GE Healthcare). The column was washed with phosphate-buffered saline (PBS) containing 40 mM imidazole and the proteins bound to the beads were eluted with a linear gradient of 40–300 mM imidazole in PBS. Aliquots of the eluate were analyzed by SDS–PAGE. Positively stained fractions were pooled and then further purified by anion-exchange chromatography as follows. The pooled fractions were applied onto a HiTrap Q column (GE Healthcare) equilibrated with 20 mM Tris–HCl pH 8.8 and eluted with a 20–500 mM linear NaCl gradient in 20 mM Tris–HCl buffer pH 8.8. The eluted fractions were collected and concentrated to 3–5 mg ml−1 using an Amicon Ultra centrifugal filter unit (30 kDa molecular-weight cutoff membrane, Millipore) for crystallization. The purity of mGluR7 LBD was analyzed by SDS–PAGE. In order to confirm the N-terminal amino acid of the secreted mGluR7 LBD, the purified protein was subjected to automated Edman degradation using an Applied Biosystems Procise model 492 protein sequencer.
2.2. Characterization of mGluR7 LBD
The oligomer formation of mGluR7 LBD was analyzed using size-exclusion chromatography and SDS–PAGE. Purified LBD protein was loaded onto a gel-filtration column (Superdex 200 PC 3.2/30, GE Healthcare) equilibrated with PBS and was eluted using the same buffer. The flow rate was 30 µl min−1 and fractions of 40 µl each were collected. Aliquots of the fractions were subjected to SDS–PAGE with a 4–20% linear gradient polyacrylamide gel (Daiichi Pure Chemicals, Japan) under reduced and nonreduced conditions followed by silver staining. Molecular-weight standards were from Daiichi Pure Chemicals, Japan.
A ligand-binding assay was performed using the polyethylene glycol (PEG) precipitation method as described previously (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶) with minor modifications. Briefly, 24 nM [3H]-labelled LY341495 ([3H]-LY341495, 1.35 TBq mmol−1, Tocris), l-glutamate at various concentrations and mGluR7 LBD protein solution were mixed in 150 µl binding buffer (40 mM HEPES pH 7.5 containing 2.5 mM CaCl2) at 277 K for 1 h. PEG 6000 was then added to the mixture to a final concentration of 15% with 3 mg ml−1 γ-globulin. After vortexing and centrifugation, the precipitated material was washed twice with 1 ml binding buffer containing 8% PEG 6000 and dissolved in 1 ml water. After the addition of 14 ml Clearsol II (Nacalai Tesque, Japan), the radioactivity was counted using a scintillation counter. The binding data were analyzed using Prism 3 (GraphPad Software). The displacement curve was fitted to a single-site binding model and the K i value was calculated.
2.3. Crystallization
Crystallization screening was performed using the hanging-drop vapour-diffusion method in 24-well VDX greased plates (Hampton Research). Initial crystallization trials were performed using Grid Screen PEG 6000 and Grid Screen Ammonium Sulfate (Hampton Research). All drops were prepared by mixing 1 µl protein solution (3–5 mg ml−1 in 20 mM Tris–HCl buffer pH 8.8 including approximately 0.2 M NaCl) on the siliconized cover glass with the same volume of reservoir solution. Protein solution prepared with 10 mM glutamate as a ligand was also used in crystallization trials. Each hanging drop was equilibrated against 0.4 ml reservoir solution at a temperature of 293 K.
2.4. Data collection
X-ray diffraction data were collected under a nitrogen-gas stream at 100 K and recorded on a CCD camera with a total oscillation range of 180° using synchrotron radiation of wavelength 1.000 Å at beamline BL41XU of SPring-8 (Hyogo, Japan). The crystals were cryoprotected by soaking briefly in the mother liquor supplemented with 15%(v/v) ethylene glycol prior to flash-freezing in a boiled-off N2 stream.
3. Results and discussion
3.1. Preparation and characterization of mGluR7 LBD
mGluR7 LBD was expressed in High Five cells and successfully secreted into the culture medium. The protein was purified to homogeneity by a combination of Ni-affinity column and anion-exchange chromatography. On analysis using SDS–PAGE followed by silver staining, the purified His-tagged LBD was observed as almost a single band (Fig. 1 ▶ b). The N-terminal amino acid of the purified protein was confirmed using a protein sequencer. Although the predicted N-terminus was at Gln35 according to the PIR database (accession No. P35400), Arg33 was detected as the first amino acid of mGluR7 LBD. This difference is probably a consequence of the inaccuracy of the prediction method. In fact, Arg33 was predicted as the N-terminal residue in the report by Okamoto et al. (1994 ▶).
Figure 1.

(a) Size-exclusion chromatography of mGluR7 LBD. The elution positions of the marker proteins are shown by arrowheads with molecular weights in kDa. The estimated molecular weight of mGluR7 LBD is 157 kDa. The indicated fractions, 1–5, were subjected to SDS–PAGE analysis as shown in Fig. 1 ▶(b). (b) Aliquots of the peaks from the gel-filtration analysis were analyzed by SDS–PAGE. Samples were run in the presence (lanes 1–5) and absence (lanes 6–10) of DTT. The gel was stained with silver nitrate. The sizes of the molecular-weight markers are shown in kDa on the left. Lanes 1 and 6, fraction 1; lanes 2 and 7, fraction 2; lanes 3 and 8, fraction 3; lanes 4 and 9, fraction 4; lanes 5 and 10, fraction 5.
It was previously reported that mGluR1 LBD formed a homodimer connected by a disulfide bond (Tsuji et al., 2000 ▶). In order to confirm the formation of the mGluR7 LBD oligomer, we used size-exclusion chromatography and SDS–PAGE analyses (Fig. 1 ▶). From size-exclusion chromatography, the estimated molecular weight of mGluR7 LBD was 157 kDa (Fig. 1 ▶ a). Because the theoretical molecular weight of the mGluR7 LBD monomer was 56 kDa, the eluted protein was definitely larger than a monomer and appeared to be a dimer or trimer. We also confirmed the oligomerization state of mGluR7 LBD by SDS–PAGE analysis. The bands of mGluR7 LBD under nonreduced conditions (Fig. 1 ▶ b, lanes 6–10) were shifted to a position corresponding to twice the molecular weight under reduced conditions (Fig. 1 ▶ b, lanes 1–5), indicating that mGluR7 LBD maintained the ability to form an interprotomer disulfide bond, as demonstrated for mGluR1 LBD (Tsuji et al., 2000 ▶).
Next, we investigated the l-glutamate-binding ability of mGluR7 LBD using [3H]-LY341495 by the previously described PEG-precipitation method (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶). The estimated K i value of 130 µM (Fig. 2 ▶) was slightly smaller than the value reported previously (869 µM; Wright et al., 2000 ▶). The result indicates that the mGluR7 LBD produced by insect cells folds correctly and that the additional His-tag residues at the C-terminus of the protein do not influence the ligand-binding capacity.
Figure 2.

Displacement of [3H]-LY341495 binding to mGluR7 LBD with l-glutamate. The indicated concentrations of glutamate with 24 nM [3H]-LY341495 were incubated with purified mGluR7 LBD (0.65 µg). Each point shows the mean ± standard deviation. The displacement curve and K i value of 130 µM (95% confidence interval = 56.9–297 µM) were obtained by sigmoidal fitting using Prism 3 (GraphPad Software).
3.2. Crystallization
mGluR7 LBD was crystallized by the hanging-drop vapour-diffusion method at 293 K. The crystals were obtained under the following three conditions: C2 of Grid Screen PEG (20% PEG 6000, 0.1 M sodium citrate buffer pH 5.0), B2 of Grid Screen Ammonium Sulfate (1.6 M ammonium sulfate, 0.1 M sodium citrate buffer pH 5.0) and C3 of Grid Screen Ammonium Sulfate (2.4 M ammonium sulfate, 0.1 M MES pH 6.0). Subsequent refinement of the buffer conditions gave an optimal crystallization condition consisting of a mixture of the B2 and C3 buffers of Grid Screen Ammonium Sulfate in a 1:4 ratio. The protein solution also included 10 mM l-glutamate. The crystals appeared within a week at 293 K and the fully grown crystals had dimensions of 0.09 × 0.06 × 0.04 mm (Fig. 3 ▶).
Figure 3.

Crystals of mGluR7 LBD. The maximum dimensions are 0.09 × 0.06 × 0.04 mm.
3.3. Data collection
For X-ray data collection, a single crystal of mGluR7 LBD picked up from a droplet using a nylon loop (Hampton Research) was transferred into cryoprotectant solution (GridScreen Ammonium Sulfate B2 and C3 in a 1:4 ratio supplemented with 15% ethylene glycol) and placed directly into a cold nitrogen-gas stream at 100 K. Diffraction data were obtained in the resolution range 100–3.30 Å (Fig. 4 ▶) and were processed using MOSFLM (Leslie, 2006 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶). The crystals belong to the trigonal space group P3121 or P3221, with unit-cell parameters a = b = 92.4, c = 114.3 Å. The asymmetric unit contains one protomer, with a solvent content of 51% and a crystal volume per protein weight V M of 2.5 Å3 Da−1. The V M value and solvent content are within the ranges usually observed for protein crystals (Matthews, 1968 ▶). The crystallographic data and data-collection statistics are summarized in Table 1 ▶. A total of 91 781 reflections containing 8884 unique data were collected with 100% completeness and an R merge of 10.6% to 3.30 Å resolution.
Figure 4.

Diffraction pattern of the crystal of mGluR7 LBD. (a) A typical 1° oscillation photograph. The concentric circles indicate resolution ranges of 10.7, 5.3, 3.6 and 2.7 Å. (b) A close-up view of the region marked by a dotted line in (a). The arrow indicates a diffraction spot at 3.3 Å resolution.
Table 1. Crystal data and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P3121 |
| Unit-cell parameters (Å) | a = b = 92.4, c = 114.3 |
| No. of protomers per ASU | 1 |
| X-ray source | BL41XU, SPring-8 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.0000 |
| Oscillation range (°) | 1.0 |
| Crystal-to-detector distance (mm) | 400 |
| No. of frames | 180 |
| Resolution range (Å) | 100–3.30 (3.48–3.30) |
| Observed reflections | 91781 |
| Unique reflections | 8884 |
| Completeness (%) | 100 (100) |
| Multiplicity | 10.3 (10.7) |
| Mean I/σ(I) | 5.4 (1.4) |
| Rmerge† | 0.106 (0.540) |
R
merge = 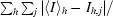
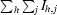 , where 〈I〉h is the mean intensity of symmetry-equivalent reflections.
, where 〈I〉h is the mean intensity of symmetry-equivalent reflections.
The structure of mGluR7 LBD has been determined by molecular replacement using the coordinates of the mGluR1 LBD structure (PDB code 1ewk; Kunishima et al., 2000 ▶) as a search probe. Calculations using AMoRe (Navaza, 1994 ▶) did not lead to structure solutions in space group P3221, but did in space group P3121. This structure determination will provide useful information for the design of mGluR group-specific drugs. For example, mGluR7 may be a potential target for the development of novel antidepressants (Cryan et al., 2003 ▶). The relationship between the determined structure and its function has been discussed elsewhere (Muto et al., 2007 ▶).
Acknowledgments
The authors would like to thank Drs N. Shimizu and T. Oyama for assistance during X-ray diffraction data collection. We are very grateful to Ms H. Kishi and Mr T. Tomura for technical support. We thank Dr S. Nakanishi for a gift of mGluR7 cDNA. The synchrotron X-ray experiment at SPring-8 was performed with the approval of the Program Advisory Committee of the Japan Synchrotron Radiation Institute (Proposal No. 2005A0339-NL1-np). This work was partly supported by a research grant endorsed by the New Energy and Industrial Technology Development Organization (NEDO).
References
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Cryan, J. F., Kelly, P. H., Neijt, H. C., Sansig, G., Flor, P. J. & van Der Putten, H. (2003). Eur. J. Neurosci.17, 2409–2417. [DOI] [PubMed] [Google Scholar]
- Hollmann, M. & Heinemann, S. (1994). Annu. Rev. Neurosci.17, 31–108. [DOI] [PubMed] [Google Scholar]
- Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., Nakanishi, S., Jingami, H. & Morikawa, K. (2000). Nature (London), 407, 971–977. [DOI] [PubMed] [Google Scholar]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Muto, T., Tsuchiya, D., Morikawa, K. & Jingami, H. (2007). Proc. Natl Acad. Sci. USA, 104, 3759–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, S. & Masu, M. (1994). Annu. Rev. Biophys. Biomol. Struct.23, 319–348. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Okamoto, N., Hori, S., Akazawa, C., Hayashi, Y., Shigemoto, R., Mizuno, N. & Nakanishi, S. (1994). J. Biol. Chem.269, 1231–1236. [PubMed] [Google Scholar]
- Suzuki, Y., Moriyoshi, E., Tsuchiya, D. & Jingami, H. (2004). J. Biol. Chem.279, 35526–35534. [DOI] [PubMed] [Google Scholar]
- Tsuchiya, D., Kunishima, N., Kamiya, N., Jingami, H. & Morikawa, K. (2002). Proc. Natl Acad. Sci. USA, 99, 2660–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, Y., Shimada, Y., Takeshita, T., Kajimura, N., Nomura, S., Sekiyama, N., Otomo, J., Usukura, J., Nakanishi, S. & Jingami, H. (2000). J. Biol. Chem.275, 28144–28151. [DOI] [PubMed] [Google Scholar]
- Wright, R. A., Arnold, M. B., Wheeler, W. J., Ornstein, P. L. & Schoepp, D. D. (2000). Naunyn Schmiedebergs Arch. Pharmacol.362, 546–554. [DOI] [PubMed] [Google Scholar]