

A recombinant mutant (C47S) of cytosolic thioredoxin peroxidase 1 from S. cerevisiae was expressed, purified and crystallized by the hanging-drop vapour-diffusion method from protein previously treated with 1,4-dithiothreitol. The crystals belong to the monoclinic space group C2 and diffraction data were collected to 2.8 Å resolution using a synchrotron-radiation source.
Keywords: cytosolic thioredoxin peroxidase 1, Saccharomyces cerevisiae
Abstract
Saccharomyces cerevisiae cytosolic thioredoxin peroxidase 1 (cTPxI or Tsa1) is a bifunctional enzyme with protective roles in cellular defence against oxidative and thermal stress that exhibits both peroxidase and chaperone activities. Protein overoxidation and/or high temperatures induce great changes in its quaternary structure and lead to its assembly into large complexes that possess chaperone activity. A recombinant mutant of Tsa1 from S. cerevisiae, with Cys47 substituted by serine, was overexpressed in Escherichia coli as a His6-tagged fusion protein and purified by nickel-affinity chromatography. Crystals were obtained from protein previously treated with 1,4-dithiothreitol by the hanging-drop vapour-diffusion method using PEG 3000 as precipitant and sodium fluoride as an additive. Diffraction data were collected to 2.8 Å resolution using a synchrotron-radiation source. The crystal structure was solved by molecular-replacement methods and structure refinement is currently in progress.
1. Introduction
Thioredoxin peroxidases (TPx) belong to a family of enzymes called peroxiredoxins that are able to reduce H2O2, peroxynitrites and a wide range of organic peroxides using a very reactive cysteinyl residue present in their active sites (Chae, Robison et al., 1994 ▶; Chae, Chung et al., 1994 ▶; Netto et al., 1996 ▶; Park et al., 2000 ▶; Nordberg & Arner, 2001 ▶; Bryk et al., 2002 ▶). In this process, the catalytic cysteine residue of TPx become oxidized. Thioredoxins and in a few cases glutaredoxins are directly involved in the reduction of TPx. While glutaredoxins are reduced by glutathione, thioredoxins are reduced by systems involving thioredoxin reductase (Trr) and nicotinamide adenine dinucleotide phosphate (NADPH; reviewed by Rouhier et al., 2001 ▶; Rouhier & Jacquot, 2005 ▶). In addition to their protective roles in oxidative or nitrosative stress, TPxs may regulate peroxide-mediated signalling cascades. Furthermore, TPx overexpression is observed in several cancers and is correlated with cellular resistance to apoptosis induced by radiation or chemotherapy-induced apoptosis (reviewed in Kang et al., 2005 ▶).
TPxs are ubiquitous proteins found from archaea to man (Park et al., 2000 ▶; Bryk et al., 2002 ▶; Herbette et al., 2002 ▶; Jung et al., 2002 ▶). In eukaryotes, TPxs can be located in the cytosol as well as in organelles such as chloroplasts, mitochondria and even in nuclei (Park et al., 2000 ▶; reviewed in Jang et al., 2006 ▶). In Saccharomyces cerevisiae, five isoforms have been described: three are cytosolic (Tsa1, Tsa2 and Ahp1), one is mitochondrial (Prx1) and another is nuclear (Dot5). The distinct or overlapping subcellular localizations create a complex network and may reflect particular physiological roles of each isoform (Park et al., 2000 ▶). In fact, yeast cells which have the Tsa1 gene deleted are specifically sensitive to oxidative stress when the mitochondrial function is inhibited (Demasi et al., 2006 ▶), accumulate aggregated proteins (Rand & Grant, 2006 ▶) and are more prone to accumulate mutations (Huang et al., 2003 ▶; Huang & Kolodner, 2005 ▶).
All TPxs use a highly conserved cysteine residue called the peroxidatic cysteine (CysP) to reduce hydroperoxides. After nucleophilic attack, the CysP is oxidized to a cysteine sulfenic acid (CysP-SOH). When TPx is exposed to high doses of peroxides, this sulfenic acid can be further oxidized to a sulfinic acid (CysP-SO2H). Initially, this overoxidized state was believed to be a dead end in the catalytic cycle, since this form is inactive and cannot be reduced by thioredoxin (Chae et al., 1999 ▶). However, Biteau et al. (2003 ▶) identified an enzyme that is able to reduce yeast sulfinic acid-overoxidized Tsa1 and Tsa2 in an ATP-dependent process.
Tsa1 and Tsa2 were characterized as homodimers of approximately 43 kDa molecular weight. Nonetheless, exposure of yeast cells to heat shock or oxidative stress triggers an intense oligomerization of Tsa1 and Tsa2 dimers, resulting in the formation of [(α2)5] and high-molecular-weight complexes (Fig. 1 ▶). This structural change is correlated with a transition of the protein function from thiol peroxidase to molecular chaperone, but the molecular mechanisms by which TPx quaternary structures assemble and dissociate are not well understood (Jang et al., 2004 ▶).
Figure 1.

Molecular switching of thioredoxin peroxidases. At low H2O2 concentrations, CysP (Cys47 in yeast Tsa1) is oxidized to CysP-SOH, which reacts with the resolving cysteine (CysR; Cys170 in Tsa1) of the other subunit in the homodimer to form an intermolecular disulfide. Under physiological conditions, the enzyme is reduced by the thioredoxin system (represented by Trx, Trr and NADPH). The peroxidase inactivation is the result of heat shock or oxidative stress. In the case of oxidative stress the overoxidation occurs at CysP (Cys47 in yeast Tsa1), resulting in CysP-SO2H. After re-establishment of homeostasis, peroxidase activity is restored by reduction of the CysP-SO2H moiety in a reaction that requires ATP hydrolysis and is catalyzed by Srx, with reducing equivalents being provided by physiological thiols (RSH) such as Trx.
Tsa1 and Tsa2 are both cytosolic enzymes that present a high primary structure homology (86% identity and 96% similarity) and exhibit peroxidatic and chaperone activities. Despite their structural and functional similarities, Ogusucu et al. (2007 ▶) showed that the pK a of the peroxidatic cysteines differ considerably between the two thioredoxin peroxidases (5.4 for Tsa1 and 6.3 for Tsa2). The difference in the peroxidatic reactivity probably relies on slight differences in the two protein structures, which may have consequences for their intrinsic peroxidatic and chaperone activities. Accordingly, human TPx cytosolic isoforms Prx1 and Prx2 (also named TPxA and TPxB, respectively), which share a very high degree of amino-acid sequence homology as in their yeast counterparts (78% identity and 91% similarity), present differences in their intrinsic chaperone and peroxidase activities. Prx1 is more efficient as a molecular chaperone, while Prx2 is a better peroxidase. A cysteine residue in Prx1 (Cys83) that is positioned at the decamer interface and is not found in Prx2 plays a critical role in these differences, since the Prx1C83S mutant presents chaperone and peroxidase activities that are very similar to those of Prx2 (Lee et al., 2007 ▶). Sequence alignment of yeast Tsa1 and Tsa2 with human Prx1 and Prx2 shows that the yeast proteins do not possess an equivalent cysteine residue but that the amino-acid residues composing the putative dimer–dimer interface differ only slightly between yeast and human TPx (Fig. 2 ▶).
Figure 2.

Alignment of the amino-acid sequences of yeast Tsa1 and Tsa2 and the human counterparts Prx1 and Prx2. The secondary-structure assignment corresponds to the human Prx2 structure. Sequence alignment was performed using ClustalW (Thompson et al., 1994 ▶). Identical residues are highlighted in green; black boxes indicate residues involved in the dimer–dimer interface in the human Prx2 structure and their equivalents in Prx1 and yeast Tsa1 and Tsa2. Black asterisks show nonconserved residues in the dimer–dimer interface region and yellow boxes show the catalytic cysteines. The red asterisk denotes Cys83 of human Prx1.
Here, we report preliminary X-ray diffraction analysis of Tsa1 from S. cerevisiae carrying the Cys47Ser substitution. The structure was solved by molecular-replacement methods using the atomic coordinates of human Prx2 (TPxB) as the search model. The comparison of the structure of Tsa1 with those of its human counterparts may help in comprehending the oligomerization process of TPx proteins and consequently in understanding the molecular mechanisms underlying their peroxidase and chaperone activities.
2. Methods
2.1. Tsa1 cloning and site-directed mutagenesis
The yeast YML028C gene encoding Tsa1 was amplified by PCR using forward primer 5′-Tsa1 (5′-CGAGAATTCACAATGGTCGCTCAA-3′) containing an EcoRI site (bold) and reverse primer 3′-Tsa1 (3′-GTTTATTCTGCGAACGGGCCCAT-5′) containing an SmaI site (bold) from genomic DNA. The PCR product was cloned into the pGEM-T Easy vector (Promega), resulting in the pGEM/tsa1 plasmid. Escherichia coli DH5α strain cells were transformed with pGEM/tsa1 and white colonies were selected from LB/ampicillin/5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-gal) medium. Plasmid extraction was performed using the Concert kit (Invitrogen). The plasmids were digested with EcoRI and SmaI, separated on a low-melting-point agarose gel, purified and introduced into pET15b expression vector. The resulting pET15b/tsa1 was transferred to E. coli BL21 (DE3) cells. Mutation of the active cysteines (Tsa1C47S and Tsa1C170S) was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene), plasmid pET15b/tsa1 and the mutagenic primers TsaMut47 (5′-TTCACTTTCGTCTCTCCAACCGAAATC-3′ and 5′-GATTTCGGTTGGACAGACGAAAGTGAA-3′) and TsaMut170 (5′-ACTGTCTTGCCACTGGACTCCA-3′ and 5′-TGGAGTCCAGTTAGATGGCAAGACAGT-3′). The plasmids pET15b/tsa1, pET15b/tsa1 C47S and pET15b/tsa1 C170S were sequenced using an Applied Biosystems ABI Prism 377 96 in order to confirm the constructions.
2.2. Expression and purification
E. coli BL21 (DE3) cells harbouring pET15b/tsa1 C47S plasmid were grown overnight in 50 ml Luria–Bertani medium containing 50 µg ml−1 ampicillin (LB/amp) at 310 K, transferred to 1 l fresh LB/amp medium and cultured further until the OD600 reached 0.6–0.8. Expression was induced with 1 mM IPTG and the cells were harvested after 4 h incubation at 310 K. The cell pellet was resuspended in 20 mM sodium phosphate pH 7.4. Cells were lysed by sonication and the cell extract was kept on ice during streptomycin sulfate (1%) treatment for 15 min. The suspension was centrifuged at 31 500g for 30 min at 277 K in order to remove nucleic acid precipitate. The resulting supernatant was filtered and applied onto a nickel affinity column (Hi-Trap, GE Healthcare). Bound protein was eluted with a linear imidazole gradient from 0 to 0.5 M. Protein purity was confirmed by SDS–PAGE. The purified protein was concentrated to 10 mg ml−1 in 5 mM Tris–HCl pH 7.5 for crystallization trials.
2.3. Crystallization and data collection
Protein samples were treated with H2O2, diamide or 1,4-dithiothreitol (DTT) (10 mM) at 310 K for 1 h and used directly in hanging-drop vapour-diffusion crystallization experiments. Initial screenings were performed at 293 K using Crystal Screen and Crystal Screen II from Hampton Research, and Wizard I and II from Jena Biosciences. Drops containing equal volumes (1.0 µl) of protein solution (10 mg ml−1 in 5 mM Tris–HCl pH 7.5) and reservoir solution were equilibrated against 0.3 ml reservoir solution. Several conditions from the initial screenings produced thin plate-shaped crystals. Promising crystals were identified in five conditions: conditions 16, 11 and 28 from Crystal Screen and conditions 16 and 31 from Crystal Screen II. All initial hits consisted of low-pH buffers with PEG as precipitant. Crystals suitable for diffraction experiments were obtained by slight variation of these conditions, such as the temperature of the assays (293 or 277 K). An additive search was also performed using Additive Screens 1, 2 and 3 from Hampton Research.
The best crystals were cryoprotected using reservoir solution supplemented with 25%(v/v) PEG 400 and cooled to 110 K in a nitrogen-gas stream. X-ray diffraction data were collected using synchrotron radiation at the D03B-MX1 protein crystallography beamline of Laboratório Nacional de Luz Síncrotron (LNLS), Campinas, Brazil. D03B-MX1 is a monochromatic beamline with maximum photon flux between 1.3 and 1.6 Å. The wavelength of the incident X-rays was set to 1.431 Å and a MAR CCD detector was used to record the oscillation data with Δϕ = 1.0°. The data set was processed using the program MOSFLM (Leslie, 1992 ▶) and the resulting intensities were scaled and merged using the program SCALA (Evans, 1993 ▶) from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Extensive attempts to determine the crystal structure of wild-type S. cerevisiae Tsa1 failed owing to the poor quality of the diffraction (maximum resolution ∼6–7 Å). We believe that partial oxidation of cysteines and/or nonspecific disulfide bridges may result in nonhomogeneous samples that yield poorly diffracting crystals. In order to overcome this problem, two approaches were adopted. Firstly, enzyme samples were treated with H2O2, diamide or DTT prior to crystallization. However, crystals obtained after this protein treatment still presented poor diffraction quality. Subsequently, two Tsa1 mutants carrying cysteine-to-serine substitutions (Tsa1C47S and Tsa1C170S) were constructed. Microcrystals of both mutants were obtained, but only Tsa1C47S produced crystals suitable for X-ray diffraction experiments.
The best Tsa1C47S crystals were obtained from a DTT-treated sample with a drop volume of 8.0 µl. 3.6 µl reservoir solution [sodium citrate pH 4.2, 10%(w/v) PEG 3000] was mixed with an equal volume of protein solution and 0.8 µl 0.1 M sodium fluoride was used as an additive. The best crystals of Tsa1C47S reached dimensions of 0.5 × 0.3 × 0.05 mm after 72 h (Fig. 3 ▶) and diffracted to 2.8 Å resolution. Crystals belong to the monoclinic space group C2, with unit-cell parameters a = 239.98, b = 51.96, c = 192.35 Å, β = 92.3°. Table 1 ▶ summarizes the data-collection statistics.
Figure 3.

Crystal of yeast Tsa1C47S obtained by hanging-drop vapour diffusion in the presence of sodium citrate pH 4.2, 10 mM sodium chloride and 10%(w/v) PEG 3000 with 100 mM sodium fluoride as an additive.
Table 1. Crystal parameters and crystallographic data statistics.
Values in parentheses are for the highest resolution shell.
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 239.98, b = 51.96, c = 192.35, β = 92.3 |
| Resolution range (Å) | 42.60–2.8 (3.2–2.8) |
| Total reflections | 230659 |
| Unique reflections | 58102 |
| Completeness (%) | 97.7 (97.7) |
| Multiplicity | 4.0 (3.8) |
| Rsym† (%) | 10.5 (42.4) |
| Average I/σ(I) | 13.7 (3.2) |
R
sym = 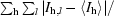
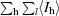 , where I
hl is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
, where I
hl is the lth observation of reflection h and 〈I
h〉 is the weighted average intensity for all observations l of reflection h.
The atomic coordinates of one monomer of the Homo sapiens Prx2 decamer (Schröder et al., 2000 ▶) were used as the search model in molecular-replacement protocols (66% sequence identity; PDB code 1qmv). The orientations and positions of the molecules in the asymmetric unit were found using MOLREP (Vagin & Teplyakov, 1997 ▶). The molecular-replacement solution showed ten monomers in the asymmetric unit and preliminary analysis of the S. cerevisiae Tsa1C47S structure revealed the quaternary organization to be similar to those observed in other previously reported crystal structures of decameric TPxs: a toroid-shaped pentamer of homodimers. Model completion and refinement are currently in progress. However, visual inspection of the initial Fourier difference maps shows that the C-terminal region of yeast Tsa1 (amino acids 167–196), which contains the resolving cysteine (Cys170), differs significantly from the human coordinates used in the molecular-replacement protocols, probably because the CysP of the human model is overoxidized to CysP-SO2H.
4. Conclusions
Here, we report the production, crystallization and preliminary X-ray analysis of the decameric form of yeast Tsa1C47S at 2.8 Å resolution. We expect that knowledge of the tertiary and quaternary structures of S. cerevisiae Tsa1 will contribute to the understanding of the molecular mechanism of peroxide decomposition and the process involved in the switch from peroxidase to chaperone activity, which may be involved in fundamental processes of cellular homeostasis. Additionally, the refined Tsa1 structure may be helpful in the construction of a good-quality theoretical model of Tsa2. It may shed light on the differences between the pK a of the peroxidatic cysteines from the two TPs cytosolic isoforms and contribute to the understanding of the molecular basis of the functional switch among thioredoxin peroxidases from different organisms.
Acknowledgments
This work was supported by grant 01/07539-5, the Structural Molecular Biology Network (SMOLBnet), from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Pesquisa e Tecnologia (CNPq), as part of the Instituto do Milênio Redoxoma, and by the Brazilian Synchrotron Light Laboratory (LNLS) under proposal D03B-MX1-5541.
References
- Biteau, B., Labarre, J. & Toledano, M. B. (2003). Nature (London), 425, 980–984. [DOI] [PubMed] [Google Scholar]
- Bryk, R., Lima, C. D., Erdjument-Bromage, H., Tempst, P. & Nathan, C. (2002). Science, 295, 1073–1077. [DOI] [PubMed] [Google Scholar]
- Chae, H. Z., Chung, S. J. & Rhee, S. G. (1994). J. Biol. Chem.269, 27670–27678. [PubMed] [Google Scholar]
- Chae, H. Z., Kim, H. J., Kang, S. W. & Rhee, S. G. (1999). Diabetes Res. Clin. Pract.45, 101–112. [DOI] [PubMed] [Google Scholar]
- Chae, H. Z., Robison, K., Poole, L. B., Church, G., Storz, G. & Rhee, S. G. (1994). Proc. Natl Acad. Sci. USA, 91, 7017–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Demasi, A. P. D., Pereira, G. A. G. & Netto, L. E. S (2006). FEBS J.273, 805–816. [DOI] [PubMed] [Google Scholar]
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Herbette, S., Lenne, C., Leblanc, N., Julien, J. L., Drevet, J. R. & Roeckel-Drevet, P. (2002). Eur. J. Biochem.269, 2414–2420. [DOI] [PubMed] [Google Scholar]
- Huang, M. E., Rio, A. G., Nicolas, A. & Kolodner, R. D. (2003). Proc. Natl Acad. Sci. USA, 100, 11529–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. E. & Kolodner, R. D. (2005). Mol. Cell, 17, 709–720. [DOI] [PubMed] [Google Scholar]
- Jang, H. H. et al. (2004). Cell, 117, 625–635. [DOI] [PubMed] [Google Scholar]
- Jang, H. H., Chi, Y. H., Park, S. K., Lee, S. S., Lee, J. R., Park, J. H., Moon, J. C., Lee, Y. M., Kim, S. Y., Lee, K. O. & Lee, S. Y. (2006). Physiol. Plant.126, 549–559.
- Jung, B. G., Lee, K. O., Lee, S. S., Chi, Y. H., Jang, H. H., Kang, S. S., Lee, K., Lim, D., Yoon, S. C., Yun, D. J., Inoue, Y., Cho, M. J. & Lee, S. Y. (2002). J. Biol. Chem.277, 12572–12578. [DOI] [PubMed] [Google Scholar]
- Kang, S. W., Rhee, S. G., Chang, T. S., Jeong, W. & Choi, M. H. (2005). Trends Mol. Med.11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W., Choi, K. S., Riddell, J., Ip, C., Ghosh, D., Park, J. H. & Park, Y. M. (2007). In the press. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Netto, L. E. S., Chae, H. Z., Kang, S. W., Rhee, S. G. & Stadtman, E. R. (1996). J. Biol. Chem.271, 15315–15321. [DOI] [PubMed] [Google Scholar]
- Nordberg, J. & Arner, E. S. (2001). Free Radic. Biol. Med.31, 1287–1312. [DOI] [PubMed] [Google Scholar]
- Ogusucu, R., Rettori, D., Munhoz, D. C., Netto, L. E. & Augusto, O. (2007). Free Radic. Biol. Med.42, 326–334. [DOI] [PubMed] [Google Scholar]
- Park, S. G., Cha, M. K., Jeong, W. & Kim, I. H. (2000). J. Biol. Chem.275, 5723–5732. [DOI] [PubMed] [Google Scholar]
- Rand, J. D. & Grant, C. M. (2006). Mol. Biol. Cell, 17, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhier, N., Gelhaye, E., Sautiere, P. E., Brun, A., Laurent, P., Tagu, D., Gerard, J., de Faÿ, E., Meyer, Y. & Jacquot, J. P. (2001). Plant Physiol.127, 1299–1309. [PMC free article] [PubMed] [Google Scholar]
- Rouhier, N. & Jacquot, J. P. (2005). Free Radic. Biol. Med.38, 1413–1421. [DOI] [PubMed] [Google Scholar]
- Schröder, E., Littlechild, J. A., Lebedev, A. A., Errington, N., Vagin, A. A. & Isupov, M. N. (2000). Structure, 8, 605–615. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Comput. Appl. Biosci.10, 19–29. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]