

The MafB transcription factor (residues 211–305) has been crystallized in complex with the 21 bp Cmare DNA-binding site. Native and mercury-derivatized data collection and preliminary analyses are reported.
Keywords: MafB, bZIP transcription factor, DNA, Cmare recognition site
Abstract
The MafB transcription factor (residues 211–305) has been overexpressed in and purified from Escherichia coli. A protein–DNA complex between the MafB homodimer and the 21 bp Maf-recognition sequence known as Cmare has been successfully reconstituted in vitro and subsequently crystallized. The diffraction properties of the protein–DNA complex crystals were improved using a combination of protein-construct boundary optimization and targeted mutagenesis to promote crystal lattice stability. Both native and mercury-derivatized crystals have been prepared using these optimized conditions. The crystals belong to space group P41212 or P43212, with unit-cell parameters a = b = 94.8, c = 197.9 Å. An anomalous difference Patterson map computed using data collected from crystals grown in the presence of HgCl2 reveals four peaks. This corresponds to two copies of the protein–DNA complex in the asymmetric unit, with a solvent content of 62% and a Matthews coefficient of 3.22 Å3 Da−1.
1. Introduction
Transcription factors are key regulators of the expression and maintenance of specific genes that together determine a particular cellular profile in a multicellular organism. This regulation process involves a dynamic network of multi-component protein–protein and protein–DNA complexes that assemble on regulatory elements of genes and either promote or inhibit transcription in response to a specific stimulus (Holstege & Young, 1999 ▶; Remenyi et al., 2004 ▶).
The Maf family of transcription factors regulate the transcription of a variety of genes during cell development and differentiation (Artner et al., 2006 ▶; Kawauchi et al., 1999 ▶; Kelly et al., 2000 ▶; Manzanares et al., 1999 ▶; Ogino & Yasuda, 1998 ▶). Characterized by sequence homology with the founding member, the v-maf oncogene, several maf-related genes have been isolated from vertebrates (Kawai et al., 1992 ▶; Nishizawa et al., 1989 ▶). Maf proteins share a set of common structural motifs that serve as a fingerprint for the family: a carboxyl-terminal leucine-zipper (bZIP) motif responsible for mediating protein homodimerization or heterodimerization, a basic DNA-binding domain and an additional ancillary domain that is not present in the canonical bZIP proteins (Kerppola & Curran, 1994 ▶). The large Maf proteins MafB, c-Maf, Nrl and MafA/LMaf additionally encompass an acidic N-terminal domain that provides transactivating activity (Nishizawa et al., 1989 ▶; Swaroop et al., 1992 ▶). The small Maf proteins MafK, MafF and MafG lack the N-terminal transactivation domain and thus may function as either transcriptional activators when heterodimerized with another bZIP protein containing transactivating activity or as negative transcriptional regulators when homodimerized (Kataoka et al., 1993 ▶; Motohashi et al., 1997 ▶).
Disruption of MafB function results in a broad range of physiological problems that reflect the variety of genes transcriptionally regulated by MafB. For example, the kreisler mouse mutant (homozygous or heterozygous MafB deletion) exhibits defects in caudal hindbrain segmentation (McKay et al., 1994 ▶). This is followed by a loss of facial motor neurons (McKay et al., 1997 ▶) and defects in the formation of the external, middle and inner ear which result in the circling behaviour after which the mouse mutant kreisler was named (Deol, 1964 ▶). A number of MafB mutations have also been associated with certain types of leukaemia (Kuehl & Bergsagel, 2002 ▶) and renal-associated diseases that result from the role of MafB in kidney podocyte differentiation (Sadl et al., 2002 ▶). Furthermore, MafB-deficient mice are reported to die shortly after birth owing to apnoea (Blanchi et al., 2003 ▶). In the haematopoietic system, MafB is an essential determinant of the cellular fate between myeloid and erythroid lineages. It is strongly upregulated in monocytes and macrophages, preventing erythroid-specific gene expression by inhibiting the activity of the transcription factor Ets-1 in myeloblasts (Sieweke et al., 1996 ▶).
Despite homology with the canonical bZIP motif, the MafB bZIP domain contains amino-acid substitutions at positions that are highly conserved in other bZIP transcription factors (Dlakic et al., 2001 ▶). This suggests that the dimerization properties of Maf proteins, although related, may confer special features that are not observed for other bZIP factors. Furthermore, Maf proteins recognize extended DNA sequences compared with the canonical bZIP members. The Maf-recognition elements (MAREs) consist of a semi-palindromic DNA sequence of 13 base pairs containing a TRE/AP-1-binding site termed Tmare [TGCTGA(C/G)TCAGCA] or a palindromic DNA sequence of 14 base pairs containing a CRE-binding site termed Cmare (TGCTGACGTCAGCA). The ancillary DNA-binding region of Maf proteins is proposed to recognize the base pairs flanking the TRE or CRE sequence core that is recognized by other bZIP proteins (Kerppola & Curran, 1994 ▶).
As part of the efforts towards structural characterization of Maf-family proteins, we here report the cloning, expression, purification and successful crystallization of a MafB construct encompassing the ancillary, DNA-binding and bZIP domains in complex with the Cmare binding site.
2. Experimental methods
2.1. Cloning and expression
The C-terminal region of MafB from Mus musculus encompassing amino acids 211–323 (MafB211–323) previously cloned into the pETM-11 expression vector (F. F. Perez) was used as a template for site-directed mutagenesis. The following primers were designed in order to introduce a Cys-to-Ser mutation (bold) at residue 298: forward, 5′-GCCTACAAGGTCAAGTCCGAGAAACTCGCCAACTCC; reverse, 5′-GGAGTTGGCGAGTTTCTCGGACTTGACCTTGTAGGC. Cys255 was not mutated to serine owing to its potential role in DNA recognition. The truncated MafB211–323,C298S construct was amplified using the forward primer 5′-CATTCCATGGGTTCCGACGACCAGCTGGTG (NcoI restriction site in bold) and the reverse primer 5′-TAAGGTACCTCAGCCGGAGTTGGCGAG (KpnI restriction site in bold). Furthermore, the forward primer 5′-CATTCCATGGGTTCCGACGACCAGCTGGTG (NcoI restriction site in bold) and reverse primer 5′-TAAGGTACCTCAGCGGCCGGAGTTGGCGAGTTTCTC were used to introduce an extra arginine residue (in bold) at the C-terminal end of the protein construct (construct MafB211–305,C298S,R306; all primers were synthesized by MWG-Biotech). The amplified PCR product was subcloned into a pETM-11 expression vector which encodes an N-terminal 6×His tag and a TEV cleavage site for subsequent affinity-tag removal by TEV protease (Parks et al., 1994 ▶). Optimal recombinant protein expression was obtained using Escherichia coli strain BL21(DE3) Codon Plus RIL (Stratagene). Cells were cultivated in LB broth medium containing 50 µg ml−1 kanamycin and 30 µg ml−1 chloramphenicol at 310 K and 190 rev min−1 until an OD600 of 0.8–0.9 was reached. The cells were then cooled to 294 K and protein expression was induced by the addition of a final concentration of 0.7 mM isopropyl β-d-thiogalactopyranoside (IPTG). After approximately 16 h incubation, cells were harvested by centrifugation (9000g, 15 min, 277 K) and stored at 253 K until further purification.
2.2. Protein purification and protein–DNA complex formation
Cell pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 8.0, 1 M urea, 100 mM NaCl, 200 mM MgCl2 and 5 mM imidazole) supplemented with 0.2%(w/v) CHAPS (Roche), 10 mM β-mercaptoethanol and 1 µg ml−1 DNaseI (Boehringer Mannheim). Cells were then lysed by lysozyme treatment (0.1 mg ml−1, 20 min on ice) followed by sonication on ice for 3 × 5 min with 30% maximum energy output (probe TT34 of the HD2200 generator from Bandelin Electronic). The resultant cell suspension was centrifuged for 45 min at 43 000g and 277 K. The clarified supernatant was then filtered (0.45 µm) and loaded onto an Ni–NTA affinity column (Qiagen) pre-equilibrated with ten column volumes (CV) of lysis buffer. After sample loading, the column was washed with 20 CV of high-salt buffer (50 mM Tris–HCl pH 8.0, 800 mM NaCl, 200 mM MgCl2, 5 mM imidazole, 3 mM β-mercaptoethanol) in order to remove any nonspecifically bound proteins, followed by a further 20 CV of washing buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 200 mM MgCl2, 40 mM imidazole, 3 mM β-mercaptoethanol). The protein was eluted with 20–30 CV of elution buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 200 mM MgCl2 and 300 mM imidazole) in a fractional manner. Immediately after elution, the protein solution was adjusted to 10 mM β-mercaptoethanol to prevent protein oxidation. Sample purity at this stage was analyzed by SDS–PAGE and the protein identification was confirmed by tandem mass spectroscopy (data not shown).
After Ni–NTA chromatography, affinity-tag removal and protein–DNA complex formation were carried out simultaneously during buffer exchange. This was performed by the addition of a final concentration of 2%(w/w) TEV protease to the protein solution together with the appropriate double-stranded oligonucleotide in a 2:1 molar ratio. 5%(v/v) glycerol was also added to the protein solution in order to avoid protein precipitation prior to DNA binding. The protein–DNA mixture was dialyzed overnight into 30 mM Tris–HCl pH 7.3, 80 mM NaCl, 50 mM MgCl2 and 3 mM β-mercaptoethanol at room temperature. The oligonucleotides containing the Cmare binding site (TAATTGCTGACGTCAGCATTA) were synthesized and purified by HPLC (Metabion). The forward and reverse single-stranded oligonucleotides were annealed (1:1 molar ratio) in 2 mM Tris–HCl pH 8.0 and 5 mM MgCl2 by incubation at 368 K for 5 min and allowed to cool slowly to room temperature.
Once the protein–DNA complex had been obtained, further purification was performed by size-exclusion chromatography using a Superdex 75 16/60 (Amersham Biosciences) column pre-equilibrated in 20 mM Tris–HCl pH 7.3, 50 mM NaCl and 3 mM DTT. Peak fractions of the protein–DNA complex were collected and analyzed by SDS–PAGE and native PAGE. Further dynamic light-scattering (DLS) analysis was performed to investigate the oligomeric state and dispersity of the complex prior to crystallization. Measurements of a minimum of 30 data points at 294 K were obtained using a DynaPro 99 instrument (Proteins Solutions Inc.) from a complex solution at 4–5 mg ml−1 and the data were analyzed using the DYNAMICS software package (Proteins Solutions Inc.).
2.3. Crystallization
The purified protein–DNA complex (5 mg ml−1) was submitted to crystallization screening trials using the hanging-drop vapour-diffusion technique and a variety of both commercially available screens and home-made sparse-matrix screens [containing polyethylene glycol (PEG) of various molecular weights (PEG 400 to PEG 8000) in buffer conditions varying from pH 4 to pH 9]. Drops containing equal volumes of protein–DNA complex solution and mother liquor (1 µl of each) were allowed to equilibrate against 700 µl reservoir solution (24-well Linbro plates) at 294 K. Heavy-atom cocrystallization experiments were performed by the addition of HgCl2 to the protein solution to a final concentration of 2 mM.
2.4. Data collection and processing
The native I diffraction data were collected on beamline X13 at EMBL/DESY, Hamburg (Germany) using a MAR CCD detector. Native II and SAD diffraction data was collected on beamline BM14 at the European Synchrotron Radiation Facility, ESRF Grenoble (France) using an ADSC Quantum-4 CCD detector. The C2 native data set was integrated in XDS (Kabsch, 1993 ▶) and scaled using the program SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Other data sets were integrated with the program MOSFLM and scaled with SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Molecular-replacement approaches were performed using the programs MOLREP (Collaborative Computational Project, Number 4, 1994 ▶) and Phaser (McCoy et al., 2005 ▶).
3. Results and discussion
3.1. Protein engineering for crystal optimization
The conserved C-terminal part of the transcription factor MafB211–305,C98S in complex with the Cmare binding site was reconstituted. Prior to crystallization experiments, the high purity of the protein–DNA complex solution was confirmed by both SDS–PAGE and native PAGE (data not shown). Although the molecular size of the protein–DNA complex could not be properly determined by gel filtration owing to its nonglobular shape, DLS could correctly estimate the molecular weight of the complex by applying the volume shape hydration of the immunoglobulin molecule as a molecular-weight model calculation. DLS analysis showed a monomodal and monodisperse distribution of the protein–DNA complex, with a calculated molecular weight of 35 kDa consistent with a 2:1 21 bp protein–DNA complex.
Optimized crystals of MafB211–305,C298S–DNA were obtained in 100 mM bis-Tris propane pH 9.0, 15%(w/v) PEG 3350 and 10%(w/v) PEG 400 (Fig. 1 ▶ a). Crystals were flash-cooled directly in a 100 K cryostream. After extensive screening of these crystals, a native data set was collected on beamline X13 at EMBL/DESY to an optical resolution of 3.1 Å (Fig. 1 ▶ and Table 1 ▶). These crystals belong to the monoclinic space group C2. Extensive molecular-replacement strategies were unsuccessful; because of both the poor reproducibility of the crystals (approximately one in 200 crystals diffracted sufficiently) and the poor quality of the diffraction (see Fig. 1 ▶ a and poor R factor), structure determination was not possible from this crystal form.
Figure 1.

(a) Monoclinic crystals (native data set I) grown in 100 mM bis-Tris propane pH 9.0, 15%(w/v) PEG 3350 and 10%(w/v) PEG 400 with a corresponding diffraction pattern collected to a resolution of 3.1 Å at beamline X13 at EMBL/DESY. (b) Mercury-derivatized tetragonal crystal (Hg SAD data set) grown in 100 mM bis-Tris propane pH 6.5, 5%(w/v) PEG 3350 and 20%(w/v) PEG 400 with a corresponding diffraction pattern collected to a resolution of 2.9 Å at BM14 at ESRF. In both figures the black bar corresponds to 50 µm.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell. Inclusion of the highly redundant data in the native II data set significantly improved the data-set completeness.
| Data set | MafB native I | MafB native II | MafB Hg SAD |
|---|---|---|---|
| Beamline | X13, EMBL/DESY | BM14, ESRF | BM14, ESRF |
| Wavelength (Å) | 0.8068 | 0.9535 | 0.9535 |
| Space group | C2 | P41212 or P43212 | P41212 or P43212 |
| Unit-cell parameters (Å, °) | a = 84.4, b = 106.6, c = 78.6, β = 91.3 | a = b = 94.8, c = 197.9 | a = b = 95.0, c = 200.1 |
| Resolution (Å) | 50–3.1 (3.37–3.10) | 36–3.1 (3.40–3.10) | 48–2.90 (3.06–2.90) |
| No. of unique reflections | 12094 | 14296 | 20968 |
| Mosaicity (°) | 2.0 | 0.6 | 0.5 |
| Redundancy | 2.6 | 7.9 | 5.0 |
| Completeness (%) | 95.4 (96.8) | 99.6 (99.6) | 99.9 (100) |
| Anomalous completeness (%) | N/A | N/A | 99.9 (100) |
| Rsym† (%) | 18.6 (34.2) | 15.3 (35.4) | 8.3 (40.8) |
| Ranom (%) | N/A | N/A | 6.1 (21.8) |
| I/σ(I) | 3.9 (2.2) | 4.6 (2.1) | 7.0 (1.8) |
R
sym = 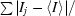
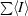 , where I
j is the observed intensity of an individual reflection and 〈I〉 is the average intensity of that reflection. R
anom is defined in the same way as R
sym except that the summation is over anomalous pairs.
, where I
j is the observed intensity of an individual reflection and 〈I〉 is the average intensity of that reflection. R
anom is defined in the same way as R
sym except that the summation is over anomalous pairs.
Encouraged by reports of protein engineering to strengthen lattice contacts within protein crystals (Derewenda, 2004 ▶), we decided to introduce an additional arginine residue at the C-terminus of the MafB211–305,C298S construct in an attempt to promote charged interactions between protein and DNA within the crystal. Extensive screening of different crystal morphologies obtained with the mutated protein led to the identification of a second crystal form with significantly improved diffraction properties. The tetragonal crystals, which grow under conditions that are almost identical except for a lower pH, are more reproducible (one in ten diffract) and have improved diffraction properties (Fig. 1 ▶ b), most notably a significant reduction in crystal mosaicity. A native data set was collected from this new crystal form on beamline BM14 at the ESRF to a resolution of 3.1 Å (Table 1 ▶). The crystals belong to point group P422. From analysis of ‘missing’ axial reflections, the actual space group is predicted to be P41212 or its enantiomorph P43212. Molecular-replacement attempts have also been unsuccessful with this second data set.
3.2. Crystal derivatization
Crystal fragility remained a problem since any kind of manipulation, such as crystal soaking, abolished the diffraction properties of the crystals. Crystal derivatization for phasing purposes was therefore attempted by heavy-atom cocrystallization. Based on the presence of two cysteines within the MafB211–305,C298S,R306 homodimer (Cys255 in each monomer), the protein–DNA complex was cocrystallized with HgCl2 and a SAD data set was collected on beamline BM14 ESRF at wavelength of 0.9535 Å, corresponding to the high-energy side of the characteristically broad Hg L III edge. The data were reduced in the tetragonal space groups P41212 or P43212, with unit-cell parameters a = b = 95.0, c = 200.1 Å. The anomalous difference Patterson map clearly suggests the presence of four mercury sites (Fig. 2 ▶), implying that there are two complexes of the MafB211–305,C298S,R306 homodimer bound to DNA in the asymmetric unit. Two protein–DNA complexes within the asymmetric unit give a Matthews coefficient of 3.22 Å3 Da−1 (Matthews, 1968 ▶) and a solvent content of 62%.
Figure 2.

Harker section (w = 0.5) of the anomalous difference Patterson map of the MafB–Cmare DNA complex cocrystallized with 2 mM HgCl2 calculated at a resolution of 4 Å using data collected at a wavelength of 0.9535 Å (Table 1 ▶) and the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶). The map is drawn with a minimum contour level of 1.5σ with 0.5σ increments. Four strong peaks observed in the Harker sections have been confirmed by analysis with SHELXD (Schneider & Sheldrick, 2002 ▶; data not shown).
4. Conclusions
Crystals of the transcription factor MafB211–305,C298S,R306 (kreisler gene) encompassing the C-terminal ancillary, DNA-binding and bZIP domains were obtained in complex with the Cmare recognition DNA motif. Protein engineering by truncation and site-targeted mutagenesis, together with minimization of crystal manipulation by introducing cryoprotectant into the crystallization conditions and cocrystallization with mercury for phasing purposes, have been key experimental approaches in obtaining reliably diffracting crystals and for derivatizing the crystal. Absolute determination of the contents of the asymmetric unit awaits structure determination.
Acknowledgments
We thank the European Synchrotron Radiation Facility and the BM14 staff for beamline support. We thank Dr Peijian Zou for useful laboratory assistance and Dr Inari Kursula and Dr Petri Kursula for help and advice during the native I data collection. This work was supported by a Volkswagen Stiftung grant (No. 1/79 996) to MW and by European Commission SPINE2 (Structural Proteomics in Europe 2) Contract No. LSHG-CT-2006-031220 to MW.
References
- Artner, I., Le Lay, J., Hang, Y., Elghazi, L., Schisler, J. C., Henderson, E., Sosa-Pineda, B. & Stein, R. (2006). Diabetes, 55, 297–304. [DOI] [PubMed] [Google Scholar]
- Blanchi, B., Kelly, L. M., Viemari, J. C., Lafon, I., Burnet, H., Bevengut, M., Tillmanns, S., Daniel, L., Graf, T., Hilaire, G. & Sieweke, M. H. (2003). Nature Neurosci.6, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Deol, M. S. (1964). J. Embryol. Exp. Morphol.12, 475–490. [PubMed] [Google Scholar]
- Derewenda, Z. S. (2004). Structure, 12, 529–535. [DOI] [PubMed] [Google Scholar]
- Dlakic, M., Grinberg, A. V., Leonard, D. A. & Kerppola, T. K. (2001). EMBO J.20, 828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege, F. C. & Young, R. A. (1999). Proc. Natl Acad. Sci. USA, 96, 2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800. [Google Scholar]
- Kataoka, K., Nishizawa, M. & Kawai, S. (1993). J. Virol.67, 2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, S., Goto, N., Kataoka, K., Saegusa, T., Shinno-Kohno, H. & Nishizawa, M. (1992). Virology, 188, 778–784. [DOI] [PubMed] [Google Scholar]
- Kawauchi, S., Takahashi, S., Nakajima, O., Ogino, H., Morita, M., Nishizawa, M., Yasuda, K. & Yamamoto, M. (1999). J. Biol. Chem.274, 19254–19260. [DOI] [PubMed] [Google Scholar]
- Kelly, L. M., Englmeier, U., Lafon, I., Sieweke, M. H. & Graf, T. (2000). EMBO J.19, 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola, T. K. & Curran, T. (1994). Oncogene, 9, 3149–3158. [PubMed] [Google Scholar]
- Kuehl, W. M. & Bergsagel, P. L. (2002). Nature Rev. Cancer, 2, 175–187. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- McKay, I. J., Lewis, J. & Lumsden, A. (1997). Eur. J. Neurosci.9, 1499–1506. [DOI] [PubMed] [Google Scholar]
- McKay, I. J., Muchamore, I., Krumlauf, R., Maden, M., Lumsden, A. & Lewis, J. (1994). Development, 120, 2199–2211. [DOI] [PubMed] [Google Scholar]
- Manzanares, M., Cordes, S., Arisa-McNaughton, L., Sadl, V., Maruthainar, K., Barsh, G. & Krumlauf, R. (1999). Development, 126, 759–769. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Motohashi, H., Shavit, J. A., Igarashi, K., Yamamoto, M. & Engel, J. D. (1997). Nucleic Acids Res.25, 2953–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa, M., Kataoka, K., Goto, N., Fujiwara, K. T. & Kawai, S. (1989). Proc. Natl Acad. Sci. USA, 86, 7711–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino, H. & Yasuda, K. (1998). Science, 280, 115–118. [DOI] [PubMed] [Google Scholar]
- Parks, T. D., Leuther, K. K., Howard, E. D., Johnston, S. A. & Dougherty, W. G. (1994). Anal. Biochem.216, 413–471. [DOI] [PubMed] [Google Scholar]
- Remenyi, A., Scholer, H. R. & Wilmanns, M. (2004). Nature Struct. Mol. Biol.11, 812–815. [DOI] [PubMed]
- Sadl, V., Jin, F., Yu, J., Cui, S., Holmyard, D., Quaggin, S., Barsh, G. & Cordes, S. (2002). Dev. Biol.249, 16–29. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Sieweke, M. H., Tekotte, H., Frampton, J. & Graf, T. (1996). Cell, 85, 49–60. [DOI] [PubMed] [Google Scholar]
- Swaroop, A., Xu, J. Z., Pawar, H., Jackson, A., Skolnick, C. & Agarwal, N. (1992). Proc. Natl Acad. Sci. USA, 89, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]