

Abstract
Nonpathogenic, resident bacteria participate in the pathogenesis of inflammation in the small intestine, but the molecular messages produced by such bacteria are unknown. Inflammatory responses involve the recruitment of specific leukocyte subsets. We, therefore, hypothesized that butyrate, a normal bacterial metabolite, may modulate chemokine secretion by epithelial cells, by amplifying their response to proinflammatory signals. We studied the expression of the chemokine, macrophage inflammatory protein-2 (MIP-2) by the rat small intestinal epithelial cell line, IEC-6. Cells were stimulated with lipopolysaccharide or with interleukin 1β (IL-1β) and incubated with sodium butyrate. Acetylation of histones was examined in Triton X acetic acid–urea gels by PAGE. Unstimulated IEC-6 cells did not secrete MIP-2. However, lipopolysaccharide and IL-1β induced MIP-2 expression. Butyrate enhanced MIP-2 secretion both in lipopolysaccharide-stimulated and IL-1β-stimulated enterocytes; but butyrate alone did not induce MIP-2 expression. Butyrate increased the acetylation of histones extracted from the nuclei of IEC-6 cells. Furthermore, acetylation of histones (induced by trichostatin A, a specific inhibitor of histone deacetylase) enhanced MIP-2 expression by cells stimulated with IL-1β. In conclusion, trichostatin A reproduced the effects of butyrate on MIP-2 secretion. Butyrate, therefore, increases MIP-2 secretion in stimulated cells by increasing histone acetylation. We speculate that butyrate carries information from bacteria to epithelial cells. Epithelial cells transduce this signal through histone deacetylase, modulating the secretion of chemokines.
Keywords: histone acetylation, chemokine, butyrate, short chain fatty acid
The primary role of the intestinal epithelium is to act as a mucosal barrier while allowing absorption of nutrients essential for life (1); however, evidence is now accumulating that the intestinal epithelium may interact with the mucosal immune system. Epithelial cells secrete cytokines, and they express class II major histocompatibility complex molecules (2–5). The small intestinal epithelium is uniquely situated between the luminal environment and cells of the mucosal immune system. Epithelial cells may, therefore, respond to changes within the intestinal lumen, by varying their signals to immune cells within the intestine (6). However, for this assertion to be valid, the epithelium should vary its response to variations in the luminal environment.
Clinically, the normal contents of the intestinal lumen play a central role in inflammatory processes in the small intestinal mucosa. Enteropathies occur in the small intestine following bacterial overgrowth. For example, in stagnant loops of bowel, bacterial populations increase when the normal motility of the intestine is deranged (7). Bacterial overgrowth also causes an enteropathy in the short bowel syndrome, where extensive resection of intestine results in an invasion of colonic bacteria (8). In addition, resident bacteria may be important in the pathogenesis of Crohn disease. Changing the diet, from a (normal) complex diet to an elemental diet, results in the remission of Crohn disease comparable to that induced by corticosteroid therapy (9). Elemental diets contain only digestible carbohydrates, amino acids, peptides, and triglycerides, with appropriate minerals and vitamins. Although many factors change within the intestine in response to an elemental diet, including food antigens and ingested nutrients, major changes are observed in the populations of nonpathogenic bacterial flora. Changes in resident bacteria may, therefore, be critical for an elemental diet to induce a remission in Crohn disease. In support of this notion is the observation that variations in normal bacterial populations directly correlate with disease activity in animal models of inflammatory bowel disease (10). However, no experiments have yet examined the mechanisms by which normal bacterial products may alter epithelial cell immune function.
Many human colonic carcinoma cell lines secrete chemokines in response to invasion by pathogenic bacteria and in response to certain cytokines (4). However, the effect of products of nonpathogenic bacteria has not been studied. It is possible that resident bacteria alter the secretion of chemokines by cells derived from the small intestine, but only in the context of other inflammatory stimuli. Macrophage inflammatory protein-2 (MIP-2) is an α-chemokine that attracts neutrophils into sites of inflammation (11–13). Macrophages secrete MIP-2, but its secretion by intestinal epithelial cells has not been reported.
Recently, Bry et al. (14) have shown that resident bacteria signal to epithelial cells inducing α1,2 fucosyl transferase, an enzyme that alters the composition of the apical glycocalyx. Introduction of Bacteroides thetaiotaomicron produced fucosylated glycoconjugates on the epithelial cell surface. However, an isogenic strain of bacteria, carrying a transposon deletion that disrupted its ability to metabolize fucose, did not induce the enzyme. The authors (14) have not yet identified the bacterial product that communicates the presence of the bacteria, but it is water soluble and not part of the bacterial structure.
We, therefore, hypothesized that metabolites of resident bacteria may modulate the secretion of MIP-2 by epithelial cells. We proposed that sodium butyrate, a major metabolite of bacteria in the intestine, may amplify the response of epithelial cells to proinflammatory stimuli. In this study, we have demonstrated that lipopolysaccharide (LPS) and interleukin 1β (IL-1β) stimulate a nonmalignant cell line (IEC-6) derived from the rat intestinal epithelium to secrete MIP-2. Butyrate enhanced this response. Butyrate also enhanced the acetylation of histones extracted from the IEC-6 cell nuclei. Independently increasing the acetylation of histone using a specific inhibitor of histone deacetylase increased the epithelial cell secretion of MIP-2 in cells stimulated with IL-1β. These experiments, therefore, have shown that butyrate amplified the secretion of MIP-2 through histone acetylation.
MATERIALS AND METHODS
Tissue Culture.
IEC-6 cells (15) were obtained from the American Type Culture Collection and plated into 12-well plates (Falcon) at an initial cell density of 5 × 104 cells per cm2. The culture media contained Dulbecco’s modified Eagle’s medium (DMEM), 10 units/ml insulin, 4 mM glutamine, 200 units/ml penicillin, and 200 μg/ml streptomycin at 37°C in 10% CO2. The media also contained 5% fetal bovine serum until day 7 after plating, when it was supplemented instead with 5 μg/ml holotransferrin (Sigma) and 5 ng/ml selenous acid (16). Butyrate (pH 7.4) (Sigma) was given 24 h before stimulation with LPS (Escherichia coli serotype 055:B5, Sigma; 10 μg/ml, Sigma) or IL-1β (1 ng/ml, R & D Systems). In most experiments, when new media were added containing LPS or IL-1β, butyrate was replaced (Fig. 1A); LPS or IL-1β was then added for a further 24 h. In experiments designed to exclude a possible direct effect of butyrate on LPS or IL-1β, butyrate was removed from the media and the cells washed twice with Hanks’ balanced salt solution before adding LPS or IL-1β (Fig. 1B). Butyrate did not significantly alter cell proliferation, as measured by cell protein concentrations at 24 and 48 h after its addition.
Figure 1.

Flow diagram describing the administration of butyrate and LPS or IL-1β. Cells were incubated in sodium butyrate for 24 h before stimulation with LPS or IL-1β. In most experiments, LPS or IL-1β was added to media containing butyrate (A). However, to ensure that there was no extracellular interaction between the LPS (or IL-1β) and butyrate, experiments were performed where butyrate was removed from the media before LPS or IL-1β (B).
Assay of MIP-2 Secretion.
The secretion of MIP-2 by IEC-6 cells was measured over 24 h. Conditioned media were collected and concentrated 15-fold by an Amicom (Centricom 3) protein concentrator according to the manufacturer’s instructions. MIP-2 was detected on Western blots using a rabbit anti-MIP-2 antibody (a gift of Barbara Sherry, Picower Institute, New York). Samples were mixed 1:2 in 2× treatment buffer (0.125 M Tris⋅HCL/4% SDS/20% glycerol/10% 2-mercaptoethanol, pH 6.8) and heated in boiling water for 3 min, before being centrifuged at 10,000 × g for 10 s to remove insoluble material. Samples (50 μl) of concentrated supernatant [and low molecular weight standards (Bio-Rad)] were loaded into a 3.5% SDS/polyacrylamide gel stacker over a SDS/15% polyacrylamide gel. After electrophoresis, samples were transferred to nitrocellulose membranes (Bio-Rad) for 90 min at 400 mA at 4°C. Nitrocellulose membranes were blocked for nonspecific binding in 5% nonfat dry milk and 0.1% Triton X-100 phosphate buffered saline (Blotto) for 2 h at room temperature and stored overnight at 4°C. The membranes were then incubated for 2 h with MIP-2 antibody at a 1:20 dilution of culture supernatant in Blotto. After washing, membranes were incubated with a 1:2,000 dilution of horseradish peroxidase-labeled donkey anti-rabbit IgG (Amersham). A second wash period (3 × 10 min) in Blotto to remove unbound antibody was followed by the application of epichemiluminescence detection reagents (Amersham) according to the manufacturer’s instructions.
Membranes were exposed to Kodak X-Omat film for autoradiography. MIP-2 was quantified by scanning the autoradiographs using a densitometer (Molecular Dynamics). MIP-2 secretion was measured by optical density of the autoradiogram and standardized by comparing each lane with a standard lane containing a cultured supernatant from LPS-stimulated RAW cells (a macrophage cell line). The concentration of this supernatant was 344 ng/ml, determined by comparing it with a quantified commercial preparation of rat MIP-2 obtained from Biosource International (Camarillo, CA). The standard was kept as a laboratory stock and was used in all experiments. Serial dilutions were used to confirm that the relationship between optical density and different concentrations of the laboratory standard was linear over the ranges used.
RNA Transfer (Northern) Blot Hybridization.
Cells were centrifuged for 3 min at 150 × g, the pellet transferred to guanidinium isothiocyanate buffer and homogenized. RNA was then isolated by phenol extraction. RNA samples (10 μg/well) were separated in 4-morpholinepropanesulfonic acid–formaldehyde agarose gels (2) and transferred to GeneScreen Plus membranes (DuPont/NEN) by capillarity. Hybridization experiments were performed using (i) MIP-2 cDNA derived from the reverse transcription of RNA collected from LPS-stimulated rat splenocytes, followed by amplification of the resulting DNA using MIP-2 specific primers. Four primers were synthesized from GenBank sequence X53798 (17): Sense: (a) GTGAACTGCGCTGTCAATGC and (b) AAGACCCTGCCAAGGGTTGA; Antisense: (c) TCGGACCTAGCATGGTCTAC and (d) GAGCGACAGACTCTCAAGTG. (All four pairs in combination gave PCR products of the predicted length.) The PCR product of the reactions initiated by primers a and d was used as a probe. This product was also cloned into a Bluescript vector and the sequence of the inserted DNA corresponded to the published sequence of rat MIP-2 (18). (ii) γ-actin cDNA (a gift of Herman Eisen, Massachusetts Institute of Technology, Cambridge, MA) (19). Inserts were labeled with [32P]dCTP (3,000 Ci/mmol; 1 Ci = 37 GBq; DuPont/NEN) with Klenow fragment after random hexanucleotide priming (20), using the PrimeIt II labeling system (Stratagene). Blots were hybridized and washed according to the manufacturer’s recommendations (DuPont/NEN). Stringent washes contained 0.5× saline sodium citrate and 0.5% SDS at 65°C. Washed blots were autoradiographed between intensifying screens at −70°C. The accumulation of MIP-2 mRNA after stimulation was expressed in comparison to γ-actin used a housekeeping gene.
Interruption of Transcription with Actinomycin D.
To examine the effects of butyrate on MIP-2 mRNA stability, transcription was interrupted with actinomycin D (10 μg/ml) 3 h after IL-1β stimulation. Enterocytes were examined before addition of IL-1β, immediately before actinomycin D (3 h), and at 6 h, 10 h, and 24 h. Results were expressed as mRNA (optical density) as a fraction of total RNA. Actin mRNA was not used as the denominator in these experiments because it also decayed after actinomycin D treatment.
Histone Extraction and Separation.
Histones were extracted from IEC-6 cells according to Cousens et al. (21). Cells were plated at 5 × 104 cells/cm2 in 75 cm2 flasks for 7 days. After 24 h incubation with either butyrate, trichostatin A (22), or media alone, cells were removed and the nuclear protein harvested by centrifuging in MLB buffer (60 mM KCl/15 mM NaCl/3 mM MgCl2/15 mM piperazine-N/N′ bis{2-ethanesulfonic acid}, pH 6.5/0.1% Nonidet P-40/0.5 mM phenylmethylsulfonyl fluoride/1 mM tetrathionate). The nuclear pellet was suspended in H2SO4 to a final concentration of 0.2 M for 2 h. The suspension was then centrifuged for 15 min and the supernatant (containing the histones) removed. The dissolved histones were precipitated with alcohol at −20°C. The precipitant was suspended in water, quantified, and 150 μg of each sample suspended in running buffer before loading onto a Triton X acetic acid–urea gel for histone separation. The gel was prepared (23) by layering an upper gel [1 M acetic acid/6.3 M urea 4.4% (wt/vol) acrylamide] onto a separating gel [1 M acetic acid/8 M urea/0.5% (vol/vol) Triton X-100/45 mM NH3/16% (wt/vol) acrylamide]. Gels were stained with Coomassie and destained, the position of the histone 4 identified with a histone 4 marker (Boehringer Mannheim).
RESULTS
Enterocytes Secrete MIP-2 in Response to LPS and IL-1β.
IEC-6 cells do not secrete MIP-2 when grown in DMEM under normal culture conditions (Fig. 2); however, the stimulation of cells with LPS (Fig. 2A) induced MIP-2 production. Thus, small intestinal epithelial cells secrete chemotactic cytokines in response to direct interaction with the bacterial product, LPS. IL-1β, a cytokine that lamina propria macrophages secrete following bacterial invasion (24), also induced MIP-2 secretion by IEC-6 cells (Fig. 2B).
Figure 2.

Effect of butyrate on MIP-2 secretion by small intestinal epithelial cells (IEC-6) induced by LPS and IL-1β. Immunoblots of 50 μl of concentrated supernatant from IEC-6 cells were probed with an anti-MIP-2 antibody detected by epichemiluminescence. Low molecular weight markers confirmed the size of the epitope-bearing protein to be 6 kDa, the weight of MIP-2. (A) Cells stimulated with LPS (10 μg/ml). (B) Cells stimulated with IL-1β (1 ng/ml). Each immunoblot is representative of three separate experiments with similar results.
Butyrate Enhances MIP-2 Secretion by Enterocytes Stimulated by LPS or IL-1β.
Butyrate alone did not stimulate MIP-2 production (Fig. 2A), but it significantly enhanced the secretion of MIP-2 after stimulation with either LPS or IL-1β (Figs. 2B and 3). This effect was due to an increased accumulation of MIP-2 mRNA in enterocytes (Fig. 4). IL-1β stimulation resulted in a peak production of MIP-2 mRNA between 3 and 4 h; butyrate not only increased the peak levels of MIP-2 mRNA, but also maintained MIP-2 mRNA accumulation after peak levels were reached (Fig. 4).
Figure 3.

Butyrate increases MIP-2 secretion from IEC-6 cells stimulated with IL-1β. Cells were incubated with butyrate (0, 1.5, and 5 mM) for 24 h before stimulation and then cultured in media containing butyrate plus IL-1β for an additional 24 h. Shown are mean and standard error of triplicate wells (representative of three experiments).
Figure 4.

Butyrate enhances MIP-2 mRNA accumulation in IEC-6 cells stimulated with IL-1β. The optical density (OD) of MIP-2 mRNA and γ-actin mRNA were measured in IEC-6 cells stimulated with IL-1β (1 ng/ml). Cells were harvested at 0, 3, 6, and 24 h after stimulation. Cells were incubated with butyrate as previously described in Fig. 1A at concentrations of 0, 1.5, and 5 mM. RNA was separated on RNA transfer (Northern) blots. Blots were incubated first with MIP-2 cDNA and then with γ-actin cDNA. Hybridization was measured after autoradiography (representative of three experiments).
To examine whether the increased MIP-2 levels were due to intracellular changes initiated by exogenous butyrate, preincubation with butyrate was followed by washing cells before adding IL-1β (Fig. 1B). This resulted in enhanced MIP-2 secretion (Fig. 5) and MIP-2 mRNA accumulation even when butyrate was absent from the medium at the time of the addition of IL-1β.
Figure 5.

Preincubation with butyrate is sufficient to induce an enhanced response of MIP-2 to IL-1β. Cells were incubated either without butyrate, with butyrate before and after stimulation (as shown in Fig. 1A), or with butyrate before IL-1β stimulation only (as shown in Fig. 1B).
Butyrate Does Not Increase the Stability of MIP-2 mRNA.
The increased accumulation of MIP-2 mRNA could be explained either by an increased stability of MIP-2 mRNA or by an increase in the transcription of the gene. Transcription was, therefore, interrupted with actinomycin D at 3 h after stimulation, when MIP-2 mRNA had reached its peak. Under these circumstances, MIP-2 mRNA decayed equally both in the cells that had received butyrate before stimulation with IL-1β and in the cells that received IL-1β alone (Fig. 6). Thus, the stability of MIP-2 mRNA was not enhanced by butyrate, and the increase in secretion was, therefore, due to increased production.
Figure 6.

Stability of MIP-2 mRNA is not altered by butyrate. Stability was examined by inhibiting transcription with actinomycin D after peak levels were reached (3 h) in cells stimulated with IL-1β in the presence and absence of butyrate (5 mM). Butyrate induced differences in MIP-2 mRNA only in those cells that continued actively to transcribe RNA (broken line). No differences were observed in mRNA after transcription had been interrupted with actinomycin D (solid line).
Butyrate Increases Histone Acetylation in IEC-6 Cells.
Butyrate is an inhibitor of histone deacetylase in many cell lines. Although butyrate is found in appreciable quantities only in the intestine, its effect on histone acetylation in intestinal epithelial cells has not been examined. Addition of 5 mM butyrate for 24 h to IEC-6 cells induced acetylation of histones, as was observed on examination of the different forms of histone 4 (Fig. 7).
Figure 7.
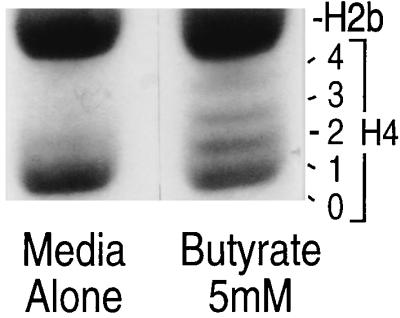
Effect of butyrate on acetylation of histone 4, displayed on Triton X acetic acid–urea polyacrylamide gel. IEC-6 cells grown in media alone contained unacetylated (0-H4) and mono-acetylated (1-H4) histone. Cells treated with 5 mM butyrate exhibit bands of higher molecular weight. They include the additional di- (2), tri- (3), and tetra- (4) acetylated forms (representative of five experiments).
Trichostatin A Induces Histone Acetylation in IEC-6 Cells and Increases MIP-2 Secretion in Response to IL-1β.
Butyrate increased both the acetylation of histones and the secretion of MIP-2 by epithelial cells when stimulated by IL-1β. If the mechanism of action of butyrate on the MIP-2 secretion were due to its ability to induce histone acetylation, then a separate method of increasing histone acetylation should similarly increase MIP-2 secretion. Trichostatin A is a specific inhibitor of histone deacetylase (22). Addition of 10 μM trichostatin A to IEC-6 cells resulted in an increase in the acetylation of histones (Fig. 8). In addition, trichostatin A added to IEC-6 cells for 24 h before stimulation of IEC-6 cells with IL-1β resulted in increased MIP-2 mRNA (Fig. 8). Trichostatin A did not affect MIP-2 mRNA accumulation in unstimulated cells (data not shown). Because of the specificity of action of trichostatin A on histone deacetylase, these data show that histone acetylation directly resulted in the increased accumulation of MIP-2 mRNA 24 h after IL-1β stimulation. Thus, the effects of butyrate on MIP-2 accumulation can be explained by its action on histone acetylation.
Figure 8.

Trichostatin A increases acetylation of histone 4 and increases MIP-2 mRNA accumulation in cells stimulated with IL-1β. Histones (Upper) and MIP-2 mRNA (Lower) were examined in the same experiment (representative of four experiments).
DISCUSSION
To the best of our knowledge, this study shows for the first time that small intestinal epithelial cells can secrete the chemokine, (MIP-2), when stimulated by IL-1β. Of particular interest, butyrate, a bacterial metabolite, increased the secretion of MIP-2 in cells stimulated by either IL-1β or LPS. Butyrate alone had no action, but it amplified the actions of these proinflammatory agents on MIP-2 secretion.
Chemotactic cytokines are classified into two groups, α- and β-chemokines, depending on the arrangement of cysteines in the center of the molecule (25). MIP-2 is an α-chemokine; it is a heparin binding protein and was originally detected in macrophages as part of their response to inflammatory stimuli (26). Many human homologues of rodent MIP-2 exist, including MIP-2α, MIP-2β, and gro (13). Like other α-chemokines, MIP-2 attracts neutrophils. Subcutaneous injection of MIP-2 into the footpads of mice elicited a localized inflammatory response (26). Moreover, intradermal injection of MIP-2 (in rabbits) induced loss of plasma from the peripheral circulation (12). If MIP-2 similarly caused a loss of plasma proteins from the intestinal vasculature, then it would induce protein-losing enteropathy, edema, and neutrophil invasion. These are the recognized features of small intestinal inflammation.
Epithelial cells and macrophages derived from the large intestine express a variety of chemokines (4, 27–30). IL-8, in particular, is induced when pathogenic bacteria invade epithelial cells (4, 31). Its secretion stimulates the chemotaxis of neutrophils in co-cultures with epithelial cells (32, 33). The ability of epithelial cells to secrete chemokines in response to inflammatory or pathogenic stimuli is well-established (4, 27). However, the contribution of the products of resident bacteria has never been examined. This article has demonstrated that butyrate increased MIP-2 secretion induced by proinflammatory agents. We have also shown (34) that butyrate enhances the secretion of IL-8 in small intestinal epithelial cells derived from the human fetus (35). In the Caco-2 epithelial cell line, butyrate increased the expression of IL-8 and GROα. However, the effects of butyrate were selective. Butyrate simultaneously decreased the secretion of the β-chemokine, monocyte chemoattractant protein-1 (36), indicating that resident bacteria may play a greater role in the recruitment of neutrophils during inflammation than they do in macrophage/monocyte recruitment.
Butyrate also alters the epithelial cell secretion of insulin-like growth factor (IGF) binding proteins (IGFBPs). Again, its effects are selective (37). Intestinal epithelial cells secrete IGFBP-2, -3, and -4 (16). Each binding protein has distinct actions on IGF bioavailability and cell proliferation. Butyrate increased the secretion of IGFBP-2, while decreasing IGFBP-3. It did not alter IGFBP-4 (37). Glutamine did not alter the profile of IGFBPs secreted. Other short chain fatty acids altered IGFBP secretion, but only in so far as they altered histone acetylation. Acetate and hexanoate, for example, did not affect IGFBP secretion. Thus, changes in IGFBP secretion were not related to the energy delivered to cells.
The concentrations of butyrate in the small intestine range from 0 to 16 mM according to estimates obtained from three different methods of sampling. First, when small intestinal contents were examined in victims of sudden accidental death, butyrate concentrations ranged from less than 1 mM in the jejunum to 1.5 mM in the ileum (38). Samples obtained from fasted children gave an estimate of 0.1 mM in the proximal small intestine (39). Finally, concentrations of 0.1–16 mM butyrate were measured in the distal small intestine sampled via ileostomies (40).
If butyrate is a normal constituent of the intestine that up-regulates MIP-2, one might ask why is the small intestine not in a state of continuous inflammation, with neutrophils attracted to the intestine. Normal, healthy small intestine contains no neutrophils. The answer to this question lies in our observation that butyrate has no action of its own (at least at the concentrations studied), but only potentiates the effects of LPS or IL-1β (Fig. 2). The healthy epithelium is not exposed to such agents in great quantities, if at all. Bacterial populations in the human small intestine are very low (only 103–104 bacteria per ml; ref. 41) and, therefore, only small amounts of LPS would reach the epithelium. During bacterial overgrowth this situation changes. Not only are there more bacteria to provide LPS, but butyrate concentrations also increase significantly (39). Stimulation by IL-1β is also not a feature of normal small intestine. IL-1β, however, is central to the pathogenesis of inflammatory bowel disease (42, 43). Under these circumstances, butyrate levels would increase the epithelial secretion of MIP-2. Reduction in butyrate by restricting nonabsorbable carbohydrate (by dietary manipulation) or eliminating bacterial populations (as seen in animal models of inflammatory bowel disease raised in germ-free conditions) would then reduce MIP-2 secretion by cells of the small intestinal epithelium.
Short-chain fatty acid enemas are of clinical benefit in patients with ulcerative colitis (44). They also reduce inflammation in patients with diverted colons (45). Such a benefit does not conflict with the data presented in this report. These diseases are characterized by epithelial cell injury. Butyrate is a nutrient of the colonic epithelial cell; it reduces epithelial permeability (46) and facilitates salt and water absorption in the colon. At early stages of ulcerative colitis, when injury to epithelial cells is greatest, the benefit of butyrate enemas is statistically significant (44). Butyrate is not used as treatment for Crohn disease, where epithelial cell damage is less evident.
This study did not examine the mechanisms by which LPS or IL-1β stimulates the enterocyte. This has been studied in many cell types (47). Furthermore, previous investigations into the effects of butyrate on intestinal epithelial cells have concentrated on its effects on cell proliferation and differentiation, particularly in epithelial cells from the large intestine (48). These include its effects on a variety of genes including: villin, the multiple drug resistance gene mdr-1 (49), the c-fos and c-myc protooncogenes (50, 51), alkaline phosphatase (52), galactosyltransferase (53), and brush border disaccharidases (54). Sodium butyrate also alters the rate of epithelial proliferation in human biopsy specimens (55). These effects are seen over a wide concentration range. However, its role in the induction of immunological genes has largely been overlooked. The possibility that butyrate may signal information from resident bacteria has not previously been considered.
In our experiments, butyrate did not induce MIP-2 secretion without an additional stimulus either from IL-1β or LPS. These data are consistent with the notion that butyrate increases the potential for transcription for the MIP-2 gene without directly initiating gene expression. It is of interest, therefore, that histone acetylation induced by trichostatin A increased MIP-2 expression when cells were stimulated with IL-1β (see Fig. 8). Trichostatin A, also, did not affect MIP-2 expression in unstimulated cells. It is well recognized that histone acetylation may increase the potential for gene expression without directly initiating transcription. This was documented in the ontogeny of chicken erythrocytes (56). In these experiments, histone acetylation was associated with the DNA in the region of the β-globin gene (which is expressed in these cells) but not with that of the albumin gene (which is not expressed in erythrocytes). However, the acetylation of β-globin histones was not temporally related to β-globin transcription: β-globin was not expressed until day 15 of fetal life, but histone acetylation was detectable in the β-globin region as early as day 3. Thus, a second signal was required for β-globin transcription. Similarly, in our experiments, butyrate (and trichostatin A) induced histone acetylation, but they did not alone induce MIP-2 secretion. Only when a second signal (LPS or IL-1β) was given did butyrate alter MIP-2 production.
Our results do not exclude the possibility that butyrate acts through a separate mechanism on the MIP-2 gene, in addition to its effects on histone acetylation. Bohan and colleagues (57) have shown that the expression of the long terminal repeats in HIV depends on the presence of SP-1 and AP-1 response elements in the long terminal repeat promoter. These investigators showed that excision of SP-1 and AP-1 sites from the plasmid in which the long terminal repeats promoter was linked to the reporter gene, chloramphenicol acetyltransferase, resulted in loss of sensitivity to butyrate. We have not performed comparable experiments with the MIP-2 promoter linked to a reporter gene to establish whether the SP-1 element in the MIP-2 protein has a similar critical role. However, we have demonstrated in electrophoretic mobility shift assays that butyrate increased the SP-1 protein available for binding to the SP-1 site (58). There is, of course, no reason nuclear protein binding and increased histone acetylation cannot coexist to alter the potential for gene transcription. Indeed, such a situation has already been demonstrated in HIV where trichostatin A increases transcription, in addition to the reported actions of SP-1 and AP-1 nuclear proteins (59).
In summary, in this study we have reported the secretion of a chemokine, MIP-2, from intestinal epithelial cells. We have shown that butyrate, a metabolite of normal bacteria, amplifies its expression. We believe that we have defined a mechanism by which resident bacteria may regulate inflammatory processes within the small intestine.
Acknowledgments
We are grateful to Dr. Barbara Sherry for the anti-MIP-2 antibody and to Dr. Robert Kingston for protocols on histone extraction and electrophoresis. This work was supported by National Institutes of Health Grants DK47753, DK40561, DK43351, and DK21474.
ABBREVIATIONS
- MIP-2
macrophage inflammatory protein-2
- IL-1β
interleukin 1β
- LPS
lipopolysaccharide
- IGF
insulin-like growth factor
- IGFBP
IGF binding protein
References
- 1.Sanderson I R, Walker W A. In: Mucosal Immunology. Ogra R, Mestecky J, McGhee J, Bienenstock J, Lamm M, Strober W, editors. San Diego: Academic; 1994. pp. 41–51. [Google Scholar]
- 2.Mason D W, Dallman M, Barclay A N. Nature (London) 1981;293:150–151. doi: 10.1038/293150a0. [DOI] [PubMed] [Google Scholar]
- 3.Mayer L, Shlien R. J Exp Med. 1987;66:1471–1483. doi: 10.1084/jem.166.5.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckmann L, Jung H C, Schurer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff M F. Gastroenterology. 1993;105:1689–1697. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- 5.Stadnyk A W. FASEB J. 1994;8:1041–1047. doi: 10.1096/fasebj.8.13.7926369. [DOI] [PubMed] [Google Scholar]
- 6.Sanderson I R. In: Nutrition in Pediatrics: Basic Science and Clinical Aspects. 2nd Ed. Walker W A, Watkins J, editors. Hamilton, ON, Canada: Decker; 1996. pp. 213–232. [Google Scholar]
- 7.Isaacs P E T, Kim Y S. Clin Gastroenterol. 1983;12:395–414. [PubMed] [Google Scholar]
- 8.Vanderhoof J A. Curr Opin Pediatr. 1995;7:560–568. doi: 10.1097/00008480-199510000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Sanderson I R, Udeen S, Davies P S W, Savage M O, Walker-Smith J A. Arch Dis Child. 1987;62:123–127. doi: 10.1136/adc.62.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rath H C, Herfarth H H, Ikeda J S, Grenther W B, Hamm T E, Balish E, Taurog J D, Hammer R E, Wilson K H, Sartor B R. J Clin Invest. 1996;98:945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolpe S D, Sherry B, Juers D, Davatelis G, Yurt R W, Cerami A. Proc Natl Acad Sci USA. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherry B, Horii Y, Manogue K R, Widmer U, Cerami A. In: Interleukin-8 (NAP-1) and Related Chemotactic Cytokines. Baggiolini M, Sorg C, editors. Vol. 4. Basel: Karger; 1992. pp. 117–130. [PubMed] [Google Scholar]
- 13.Widmer U, Manogue K R, Cerami A, Sherry B. J Immunol. 1993;150:4996–5012. [PubMed] [Google Scholar]
- 14.Bry L, Falk P G, Midtvdet T, Gordon J I. Science. 1996;273:1380–1383. doi: 10.1126/science.273.5280.1380. [DOI] [PubMed] [Google Scholar]
- 15.Quaroni A, Wands J, Treldstad R L, Isselbacher J K. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oguchi S, Walker W A, Sanderson I R. Am J Physiol. 1994;267:G843–G850. doi: 10.1152/ajpgi.1994.267.5.G843. [DOI] [PubMed] [Google Scholar]
- 17.Tekamp-Olson P, Gallegos C, Bauer D, McClain J, Sherry B, Fabre M, van Deventer S, Cerami A. J Exp Med. 1990;172:911–919. doi: 10.1084/jem.172.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Driscoll K E, Hassenbein D G, Howard B W, Isfort R J, Cody D, Tindal M H, Suchanek M, Carter J M. J Leukocyte Biol. 1995;58:359–364. doi: 10.1002/jlb.58.3.359. [DOI] [PubMed] [Google Scholar]
- 19.Enoch T, Zinn K, Maniatis T. Mol Cell Biol. 1986;6:801–810. doi: 10.1128/mcb.6.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feinberg A P, Vogelstein B. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- 21.Cousens L S, Alberts B M. J Biol Chem. 1982;257:3945–3949. [PubMed] [Google Scholar]
- 22.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 23.Arts J, Lansink M, Grimbergen J, Toet K H, Kooistra T. Biochem J. 1995;310:171–176. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Issekutz A C, Megyeri P, Issekutz T B. Lab Invest. 1987;56:49–59. [PubMed] [Google Scholar]
- 25.Baggiolini M, Dewald B, Moser B. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 26.Wolpe S D, Davatelis G, Sherry B, Beutler B, Hesse D G, Nguyen H T, Moldawer L L, Nathan C F, Lowry S F, Cerami A. J Exp Med. 1988;167:570–581. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson P, Rosella O. Gut. 1995;37:536–543. doi: 10.1136/gut.37.4.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grimm M C, Elsbury S K, Pavli P, Doe W F. Gut. 1996;38:90–98. doi: 10.1136/gut.38.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Izutani R, Loh E Y, Reinecker H-C, Ohno Y, Fusunyan R D, MacDermott R P. Inflamm Bowel Dis. 1995;1:37–47. [PubMed] [Google Scholar]
- 30.Reinecker H-C, Loh E Y, Ringler D J, Mehta A, MacDermott R P. Gastroenterology. 1995;108:40–50. doi: 10.1016/0016-5085(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 31.McCormick B A, Colgan S P, Delp-Archer C, Miller S I, Madara J L. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly C P, Keates S, Siegenberg D, Linevsky J K, Pothoulakis C, Brady H R. Am J Physiol. 1994;267:G991–G997. doi: 10.1152/ajpgi.1994.267.6.G991. [DOI] [PubMed] [Google Scholar]
- 33.McCormick B A, Hofman P M, Kim J, Carnes D K, Miller S I, Madara J L. J Cell Biol. 1995;131:1599–1608. doi: 10.1083/jcb.131.6.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fusunyan R D, MacDermott R P, Walker W A, Sanderson I R. Pediatr Res. 1995;37:122A. (abstr.). [Google Scholar]
- 35.Sanderson I R, Ezzell R M, Kedinger M, Erlanger M, Xu Z, Pringault E, Leon-Robine S D, Walker W A. Proc Natl Acad Sci USA. 1996;93:7717–7722. doi: 10.1073/pnas.93.15.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinn J J, Fusunyan R D, MacDermott R P, Sanderson I R. Gastroenterology. 1996;110:A833. (abstr.). [Google Scholar]
- 37.Nishimura A, Martinez O, Oguchi S, Fusunyan R D, MacDermott R P, Sanderson I R. Gastroenterology. 1995;108:A742. (abstr.). [Google Scholar]
- 38.Cummings J H, Pomare E W, Branch W J, Naylor C P E, MacFarlane G T. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lloyd D R, Brown J D, Brown G A, Booth I W. Acta Paediatr. 1992;81:51–56. doi: 10.1111/j.1651-2227.1992.tb12078.x. [DOI] [PubMed] [Google Scholar]
- 40.Nasmyth D G, Godwin P G R, Dixon M F, Williams N S, Johnston D. Gastroenterology. 1989;96:817–824. [PubMed] [Google Scholar]
- 41.Marteau P, Pochart P, Bouhnik Y, Rambaud J-C. In: Intestinal Flora, Immunity, Nutrition and Health. Simopoulos A P, Corring T, Rerat A, editors. Basel: Karger; 1993. pp. 1–21. [Google Scholar]
- 42.Cominelli F, Nast C C, Clark B D, Schindler R, Lierena R, Eysselein V E, Thompson R C, Dinarello C A. J Clin Invest. 1990;86:972–980. doi: 10.1172/JCI114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Youngman K R, Simon P L, West G A, Cominelli F, Rachmilewitz D, Klein J S, Fiocchi C. Gastroenterology. 1993;104:749–758. doi: 10.1016/0016-5085(93)91010-f. [DOI] [PubMed] [Google Scholar]
- 44.Breuer R I, Soergel K H, Lashner B A, Christ M L, Hanauer S B, Vanagunas A, Harig J M, Keshavarzian A, Robinson M, Sellin J H, Weinberg D, Vidican D E, Flemal K L, Rademaker A W. Gut. 1997;40:485–491. doi: 10.1136/gut.40.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harig J M, Soergel K H, Komorowski R A, Wood C M. N Engl J Med. 1989;320:23–28. doi: 10.1056/NEJM198901053200105. [DOI] [PubMed] [Google Scholar]
- 46.Vermia P, Cittadini M, Caprilli R, Torsoli A. Dig Dis Sci. 1995;40:305–308. doi: 10.1007/BF02065414. [DOI] [PubMed] [Google Scholar]
- 47.Beg A A, Finco T S, Nantermet P V, Baldwin A S. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bugaut M, Bentejac M. Annu Rev Nutr. 1993;13:217–241. doi: 10.1146/annurev.nu.13.070193.001245. [DOI] [PubMed] [Google Scholar]
- 49.Petit J M, Chauffert B, Dimanche-Boitrel M T, Genne P, Duchamp O, Martin F. Anticancer Res. 1993;13:487–490. [PubMed] [Google Scholar]
- 50.Souleimani A, Asselin C. Biochem Biophys Res Commun. 1993;193:330–336. doi: 10.1006/bbrc.1993.1628. [DOI] [PubMed] [Google Scholar]
- 51.Taylor C W, Kim Y S, Childress-Fields K E, Yeoman L C. Cancer Lett. 1992;62:95–105. doi: 10.1016/0304-3835(92)90179-y. [DOI] [PubMed] [Google Scholar]
- 52.Herz F. Biochim Biophys Acta. 1993;1180:289–293. doi: 10.1016/0925-4439(93)90052-3. [DOI] [PubMed] [Google Scholar]
- 53.Shah S, Lance P, Smith T J, Berenson C S, Cohen S A, Horvath P J, Lau J T Y, Baumann H. J Biol Chem. 1992;267:10652–10658. [PubMed] [Google Scholar]
- 54.Gibson P R, Moeller I, Kagelari O, Folino M, Young G P. J Gastroenterol Hepatol. 1992;7:165–172. doi: 10.1111/j.1440-1746.1992.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 55.Friedel D, Levine G M. J Parenter Enteral Nutr. 1992;16:1–4. doi: 10.1177/014860719201600101. [DOI] [PubMed] [Google Scholar]
- 56.Hebbes T R, Clayton A L, Thorne A W, Crane-Robinson C. EMBO J. 1994;13:1823–1830. doi: 10.1002/j.1460-2075.1994.tb06451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bohan C A, Robinson R A, Luciw P A, Srinivasan A. Virology. 1989;172:573–583. doi: 10.1016/0042-6822(89)90200-6. [DOI] [PubMed] [Google Scholar]
- 58.Ohno Y, Fusunyan R D, Aoki N, MacDermott R P, Sanderson I R. Gastroenterology. 1995;108:A887. (abstr.). [Google Scholar]
- 59.Van Lint C, Emiliani S, Ott M, Verdin E. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]