

Abstract
Transthyretin (TTR) is normally a stable plasma protein. However, in cases of familial TTR-related amyloidosis and senile systemic amyloidosis (SSA), TTR is deposited as amyloid fibrils, leading to organ dysfunction and possibly death. The mechanism by which TTR undergoes the transition from stable, soluble precursor to insoluble amyloid fibril and the factors that promote this process are largely undetermined. Most models involve the dissociation of the native TTR tetramer as the initial step. It is largely accepted that the TTR gene mutations associated with TTR-related amyloidosis lead to the expression of variant proteins that are intrinsically unstable and prone to aggregation. It has been suggested that amyloidogenicity may be conferred to wild-type TTR (the form deposited in SSA) by chemical modification of the lone cysteine residue (Cys10) through mixed disulfide bonds. S-Sulfonation and S-cysteinylation are prevalent TTR modifications physiologically, and studies have suggested their ability to modulate the structure of TTR under denaturing conditions. In the present study, we have used fluorescence-detected sedimentation velocity to determine the effect of S-sulfonate and S-cysteine on the quaternary structural stability of fluorophore-conjugated recombinant TTR under nondenaturing conditions. We determined that S-sulfonation stabilized TTR tetramer stability by a factor of 7, whereas S-cysteinylation enhanced dissociation by 2-fold with respect to the unmodified form. In addition, we report the direct observation of tetramer stabilization by the potential therapeutic compound diflunisal. Finally, as proof of concept, we report the sedimentation of TTR in serum and the qualitative assessment of the resulting data.
The amyloidoses are a class of protein folding disorders in which one of over 20 different soluble precursor proteins misfolds, aggregates, and deposits in various tissues, causing organ dysfunction and, in many cases, death (1–3). One of these precursor proteins is transthyretin (TTR3; Swiss-Prot accession number P02766), a small (Mr 13761) plasma protein that self-associates into tetramers under physiological conditions. Normally a stable protein, TTR has been implicated in two distinct forms of systemic amyloidosis, familial TTR-related amyloidosis (ATTR) and senile systemic amyloidosis (SSA). ATTR usually involves a point mutation in the coding region of the TTR gene, leading to the production of a variant protein containing one of over 80 pathological amino acid substitutions (4). Amyloid deposits in ATTR generally manifest clinically as polyneuropathies and/or cardiomyopathies, depending on the organs of involvement (5). In SSA, wild-type TTR deposition occurs in the myocardium (6) and can cause atrial fibrillation and congestive heart failure (7–11). Pathological studies have estimated that ∼25% of individuals over the age of 80 may be afflicted with SSA (9), and as the elderly population in industrialized countries continues to grow, the disease will probably contribute a significant yet underrecognized source of cardiac dysfunction. It is, therefore, of great importance to establish a molecular understanding of the factors promoting the disease and to investigate strategies whereby these processes may be abrogated.
In both SSA and ATTR, the mechanism underlying TTR structural rearrangement, aggregation, and deposition is largely undetermined. However, in recent years, in vitro studies have suggested that perturbation of the native structure is required for (or at least accelerates) amyloid formation (12, 13). Under denaturing conditions, tetramer dissociation is the initial and rate-limiting step of fibrillogenesis (14–16). Under physiological conditions, ATTR-related amino acid substitutions probably enhance the population of monomeric, aggregation-prone subunits either by directly destabilizing the tetramer (quaternary structural destabilization) or by lowering the free energy of the partially unfolded monomer (tertiary/secondary structural destabilization). It is not immediately obvious how structural instability could be conferred to wild-type TTR, but post-translational modifications are a possibility. In human serum, TTR is extensively modified at its lone cysteine residue (Cys10) through mixed disulfide bonds with a number of different compounds (17–20). Of these, adducts of sulfonate (S-sulfonation) and cysteine (S-cysteinylation) are generally the most common. Several research groups, including ours, have hypothesized that Cys10 modifications modulate TTR stability at the quaternary or tertiary/secondary level, thereby altering the propensity of wild-type TTR to form amyloid fibrils.
Much insight into the structural basis of TTR amyloid formation has been realized in recent years with techniques such as chaotrope-, thermal-, and acid-mediated unfolding assays. These methods provide a useful paradigm for analyzing differential structural effectors, such as amino acid substitutions or post-translational modifications, between related samples under denaturing conditions but do not allow straightforward generalization to physiological conditions. These methods are practical, because, with few exceptions (21, 22), neither wild-type nor variant TTR aggregate at physiological pH within an experimentally practical time frame. However, the critical step in fibrillogenesis, tetramer dissociation, could theoretically be evaluated by primary analytical methods under nondenaturing conditions. Since all previous evidence suggests the importance of this step, it is desirable to provide first principles methods for evaluating effectors of tetramer stability.
Sedimentation velocity analytical ultracentrifugation is a powerful method for analyzing protein self-association (23–25). One of the more general approaches for the analysis of such systems is the determination of the isotherm of weight-average sedimentation coefficients (s̄) as a function of concentration (26–29). The concentration dependence of s̄ is a thermodynamic phenomenon governed solely by the law of mass action, and the experimental measurement in the ultracentrifuge is completely independent of solvent composition. An experimental limitation is that the molecule must be optically detectable at concentrations where dissociation occurs. Therefore, the sensitivity limits of the standard centrifuge optical systems may restrict the analysis of tightly associating systems. Such is the case with TTR where, for the wild-type protein, dissociation is not detected at concentrations accessible with either the absorbance or interference optical systems. Recently, fluorescence detection has been developed for the analytical ultracentrifuge (30). The superior sensitivity of this system allows detection at concentrations several orders of magnitude lower than the existing optical systems. In addition, the selectivity of fluorescence allows the detection of target molecules contained in complex biological fluids like plasma, urine, or cerebrospinal fluid.
In the current study, fluorescence-detected sedimentation velocity was used to examine the contribution of two biologically relevant cysteine modifications to the stability of the TTR tetramer under nondenaturing conditions. These conditions effectively uncouple quaternary structural perturbations from tertiary and secondary effects, a differentiation not allowed by the previously employed denaturing techniques. In addition, the effect of diflunisal, a well characterized stabilizer of the TTR tetramer (31, 32), on the s̄ isotherm has been investigated. Finally, the possibility of using this technique directly in a complex biological fluid has been investigated by analyzing the sedimentation profile of fluorescein-labeled TTR in human serum.
EXPERIMENTAL PROCEDURES
Reagents—Fluorescein-5-isothiocyanate (FITC “Isomer I”) was purchased from Invitrogen. Diflunisal (5-(2,4-difluorophenyl)-2-hydroxybenzoic acid), ovalbumin, and DMSO were purchased from Sigma. All other reagents were from Fisher and were of the highest quality available. Human serum was obtained with institutional approval, in accordance with current institutional review board protocols.
Protein Sample Preparation—Recombinant human transthyretin (rTTR) was prepared and S-sulfonated (rTTR-SO3H) or S-cysteinylated (rTTR-Cys) as previously described (33). Each preparation demonstrated >95% homogeneity within experimental error (∼4 units) of the expected molecular weight (rTTR, Mr 13761; rTTR-SO3H, Mr 13841; rTTR-Cys, Mr 13880) as assessed by electrospray ionization mass spectrometry (ESI-MS) (see below). The three recombinant proteins were then chemically coupled to the amine-reactive 5-isothiocyanate derivative of fluorescein (FITC). Solutions of 5 mg of each protein were exchanged into 0.1 m sodium bicarbonate, pH 9, using EconoPak 10-DG desalting columns (Bio-Rad), and the volumes were adjusted to 3 ml (final concentration, 1.7 mg/ml). FITC was dissolved in dimethyl formamide at 10 mg/ml, and 100 μl was added to each protein solution. The reaction mixtures were incubated for 1 h at room temperature. The unreacted free dye was removed, and the samples were exchanged into 100 mm potassium chloride, 10 mm Tris, pH 7.5, using EconoPak 10-DG desalting columns.
Concentrations were determined by the absorbance at 280 nm using E1%280, 14.1 ml mg–1 cm–1 (34). The signal was corrected for the absorbance of the dye by the following equation,
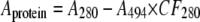 |
(Eq. 1) |
where Aprotein is the corrected protein absorbance at 280 nm, A280 is the measured absorbance of the conjugate at 280 nm, A494 is the measured absorbance of the conjugate at 494 nm, and CF280 is the correction factor, 0.30, as tabulated for fluorescein by Invitrogen. Samples were diluted to 0.5 mg/ml and stored at –20 °C until use. Sample quality and protein/dye stoichiometry were determined by ESI-MS and liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described below. Molar concentrations are reported in reference to the physiologic tetrameric species.
For sedimentation analysis of FITC-labeled rTTR species (F-rTTR, F-rTTR-SO3H, and F-rTTR-Cys), stock solutions (0.5 mg/ml) were diluted to 6.25 μg/ml in 0.1 mg/ml ovalbumin, 100 mm potassium chloride, 10 mm Tris, pH 7 or 9. A 2-fold dilution series was then constructed, using the same buffer, over the range 6.25 μg/ml to 12.2 ng/ml. For analysis of drug binding, diflunisal was dissolved to 10 mg/ml in DMSO and then diluted 1:100 in 0.1 mg/ml ovalbumin, 100 mm potassium chloride, 10 mm Tris, pH 9, and used for a 2-fold dilution series of F-rTTR over the protein concentration range 6.25 μg/ml to 48.8 ng/ml. For the control, DMSO was used instead of the stock diflunisal solution. For measurement of sedimentation in serum, stock F-rTTR was diluted to 6.25 μg/ml in human serum. All samples were equilibrated for ∼1 h at room temperature and then ∼1 h in the centrifuge vacuum chamber at experimental rotor temperature before sedimentation was initiated.
ESI-MS—Samples were prepared for mass spectrometry by reversed phase HPLC. Approximately 5 μg of each sample was loaded onto a Zorbax Poroshell 300SB-C8 HPLC column (75 mm long, 2.1-mm inner diameter, 5-μm particle size) that was pre-equilibrated in 95% solvent A (5% acetonitrile, 0.1% trifluoroacetic acid in water) and 5% solvent B (0.085% trifluoroacetic acid in acetonitrile). The column was eluted with a 5-min linear increase in solvent B to 85% at 1 ml/min. The entire protein peak, as indicated by A210, was collected, dried in a centrifugal concentrator (ThermoSavant, Holbrook, NY), and stored at –20 °C until analysis.
The samples were dissolved in 10 μl of 50% acetonitrile, 0.1% formic acid in water and loaded into 1-μm aperture nanospray tips prepared in house using a Sutter (Novato, CA) capillary puller and 1.2 × 76-mm thin wall borosilicate glass capillaries (World Precision Instruments, Sarasota, FL). The samples were introduced into a Micromass Quattro II triple quadrupole mass spectrometer by continuous nanospray generated with the following instrument settings: ion source block temperature, 120 °C; capillary potential, ∼1200 V; cone potential, 32 V; extractor potential, 7 V; RF lens potential, 0.2 V. The raw data, collected over the range m/z 200–1950, were deconvoluted (35) using the MAXENT function of the MASSLYNX (version 3.4) software.
The protein/dye stoichiometries of the F-rTTR conjugates were determined by the following equation,
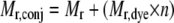 |
(Eq. 2) |
where Mr,conj is the neutral mass of the conjugate determined experimentally, Mr is the neutral mass of the initial species (rTTR, 13761; rTTR-SO3H, 13841; rTTR-Cys, 13880), Mr,dye is the neutral mass of FITC (389, with no change in mass upon coupling), and n is the number of dye molecules bound per molecule of initial species.
Tryptic Digestion/LC-MS/MS—Reversed phase HPLC-purified F-rTTR (2.4 μg) was incubated with a 1:100 enzyme-to-substrate ratio of trypsin gold, mass spectrometry grade (Promega, Madison, WI) in 0.1 m ammonium bicarbonate, pH 8, for 17 h at 37 °C. The reaction was neutralized with 1 μl of 10% formic acid and dried in a centrifugal concentrator. Dried samples were resuspended at ∼2 pm in 3% acetonitrile in water and analyzed with a LTQ-Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with a Waters NanoAcquity Ultra Performance LC (Waters Corp., Milford, MA) equipped with a 100 μm × 10 cm C18 capillary. The capillary was equilibrated at 97% solvent C (0.1% trifluoroacetic acid in water), 3% solvent D (0.1% trifluoroacetic acid in acetonitrile) prior to sample application. After loading, peptides were eluted from the capillary at 500 nl/min with a linear increase in solvent D from 3 to 60% over 37 min, followed by a second increase from 60 to 80% over 10 min. Continuous electrospray was initiated at 140 V with the heated capillary set at 320 °C. For MS/MS analyses, each ion of interest was isolated with an isolation window of 3.0 Da and fragmented by collision-induced dissociation with normalized collision energy of 35%.
Circular Dichroism Spectroscopy—Circular dichroism spectroscopy experiments were conducted with an Aviv 215 spectropolarimeter (Aviv Biomedical, Lakewood, NJ) equipped with a nitrogen-purged sample holder held at 25 °C with a thermoelectric temperature controller. Samples (12 μm in 10 mm Tris, 100 mm KCl, pH 7 or 9) were loaded into 1-mm path length quartz cuvettes for far UV experiments or 5-mm path length quartz cuvettes for near UV experiments. Data were collected in 1-nm increments (1-nm band width) over the spectral range of 190–250 nm (far UV experiments) and 250–320 nm (near UV experiments) with an averaging time of typically 20–30 s. The CD spectra were presented in ellipticity units per mole of residue for far UV experiments and per mole of protein for near UV experiments. Far UV spectra were smoothed using the Aviv noise reduction routine.
Analytical Ultracentrifugation—Sedimentation velocity experiments were conducted with an Optima XLI analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) retrofitted with a prototype fluorescence optical system (commercial version available from Aviv Biomedical). Samples were loaded into ultracentrifuge cells containing graphite-filled epoxy SedVel60K sedimentation velocity centerpieces (Spin Analytical, Durham, NH) and fused silica optical windows. The centrifuge and data acquisition were controlled with the Advanced Operating System (AU-AOS). For all experiments, samples were centrifuged in an An50-Ti rotor at 50,000 rpm with the temperature held at 20 °C. Data were acquired at 20-μm radial increments, averaging 5 revolutions/scan. The photomultiplier gain and the programmable gain amplifier were set for each cell to minimize noise while providing adequate signal for data analysis (>100 fluorescence units, determined visually and set at 3,000 rpm).
Analysis of Sedimentation Data—Sedimentation velocity data were analyzed using the program SEDFIT (version 9.4) (36) (available on the World Wide Web). The s̄ value of each data set was determined in two steps. First, the continuous c(s) distribution model (36, 37) with 68% confidence level was used to generate the best fit of the data. In addition, an estimate of s̄ was obtained by integration of the resulting distribution (36). Second, using the best fit parameters from the c(s) distribution, the Monte Carlo simulation routine of the analysis package was used to calculate s̄ and the associated error interval.
The signal to noise ratio (S/N) of each data set was calculated by the following equation,
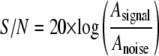 |
(Eq. 3) |
where Asignal is the amplitude of the fluorescence signal determined by subtracting the base-line signal from the plateau signal of the first scan, and Anoise is the amplitude of the time- and position-invariant noise determined by the r.m.s.d. of the finite element fit.
For modeling of isotherm data, the s̄ values determined in SEDFIT were converted to standardized s̄20, W values (s̄ in water at 20 °C) using the program SEDNTERP (version 1.08, available on the World Wide Web). In these calculations, buffer viscosity (ηB) and density (ρB) at 20 °C (experimental temperature) were estimated from composition: ηB = 0.01469 poise, ρB = 1.00325 g/ml. For water, standard parameters were assumed: ηW = 0.01002 poise, ρW = 0.99823 g/ml. The partial specific volume (v̄) of TTR was estimated as 0.7324 ml/g (at 20 °C) from composition. The s̄20, W isotherms were modeled using the program SEDPHAT (28) (version 4.4b; available on the World Wide Web). Models for monomer-tetramer (1→4) and monomerdimer-tetramer (1→2→4) self-association were applied and assessed for appropriateness by the F-statistic,
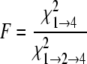 |
(Eq. 4) |
where 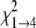 and
and
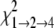 are the χ values for
the fits to the two respective models. In each case, the calculated value was
compared with the value of F = 3.39 (α = 0.05) tabulated for 9
degrees of freedom (df) for the 1→4 model and 8 df for
the 1→2→4 model, which were calculated as follows,
are the χ values for
the fits to the two respective models. In each case, the calculated value was
compared with the value of F = 3.39 (α = 0.05) tabulated for 9
degrees of freedom (df) for the 1→4 model and 8 df for
the 1→2→4 model, which were calculated as follows,
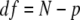 |
(Eq. 5) |
where N is the number of data points (10 for both models), and p is the number of fitting variables (1 for the 1→4 model and 2 for the 1→2→4 model).
In all cases, the following fitting parameters were constrained: s1, 1.8 S; s2 (in the 1→2→4 model), 2.7 S; s4, 4.6 S. Additionally, M1 values were set to the Mr determined by ESI-MS and constrained during the fit. The parameter for 4→1 dissociation reported herein, c½, is the concentration of half-dissociation and is related to the dissociation constant (KD) by the following equation,
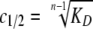 |
(Eq. 6) |
where n is the stoichiometry of the self-association.
For the analysis of sedimentation in serum, s was determined qualitatively from the slope of a plot of the natural log of the relative radial boundary position (ln r/rm) as a function of the angular velocity squared multiplied by the time of the given scan (ω2t). The radial boundary position (r) was determined as the half-height of the plateau region for each scan, whereas the meniscus position (rm) was determined visually and used as a constant for all analyzed scans. In addition, s was determined from the continuous c(s) distribution (36, 37) as described above.
RESULTS
TTR in its native state is tetrameric. According to most models of TTR fibrillogenesis, only monomeric TTR misfolds into amyloid fibrils. Consequently, the TTR tetramer must dissociate prior to amyloid formation. To investigate the effect of biologically relevant cysteine adducts on TTR tetramer stability and their potential role as modulators of amyloid formation, fluorescent derivatives of unmodified, S-sulfonated, and S-cysteinylated rTTR were prepared, and self-association isotherms were determined by fluorescence-detected analytical ultracentrifugation. In vivo, TTR tetramers are probably composed of a mixture of Cys10-modified species. However, to simplify the interpretation of the data, homotetramers of each form were studied.
Each form of rTTR was coupled to FITC, an amine-reactive form of fluorescein. TTR contains nine potential conjugation sites (primary amines) for FITC per monomer (eight lysine residues and the N-terminal glycine residue). This allows a significant possibility for sample heterogeneity, which may complicate interpretation of dissociation. To characterize the heterogeneity introduced by fluorescent probe derivatization, several different mass spectrometric approaches were employed. ESI-MS was conducted on each fluorescent derivative, and in each case, a single predominant species was identified with an observed neutral mass consistent with the mass expected for a product with a 1:1 dye/monomer stoichiometry (F-rTTR, Mr 14150; F-rTTR-SO3H, Mr 14230; F-rTTR-Cys, Mr 14270). Minor species corresponding to unconjugated as well as 2:1 dye/monomer stoichiometry were also observed in all three cases, although these impurities were estimated to account for <15% of the total sample preparation. In addition, a minor species of Mr 14230 was observed in the F-rTTR sample, possibly indicating the presence of a small amount of S-sulfonation or other contaminant. The conjugation site specificity of the 1:1 dye/monomer product was determined by LC-MS/MS analysis of a tryptic digestion of F-rTTR. An abundant ion corresponding to the mass of the derivatized N-terminal tryptic peptide (FITC-Gly1-Lys9) was observed at m/z 611.72 [M + 2H]2+ (calculated m/z 611.72 [M + 2H]2+). The most abundant product ions observed in the MS/MS spectrum of this precursor ion were m/z 390.10 and m/z 833.45, which can be assigned as the protonated free label (calculated m/z 390.04 [M + H]+) and underivatized N-terminal tryptic peptide (calculated m/z 833.40 [M + H]+), respectively. The observance of these ions is consistent with reports that the fluorescein adduct is labile under collisional activation conditions (38). Although the MS/MS spectrum did not contain a fragment that specifically located the site of conjugation, observance of the FITC-Gly1–Lys9 tryptic peptide provides sufficient evidence to confidently assign the primary labeling site to the N-terminal glycine residue, since modification of Lys9 would have prevented tryptic digestion at this site (39). The plot of m/z 390.0, corresponding to the signal from the fluorescein fragment ion, did not show any other maxima during the chromatographic elution, indicating that none of the other tryptic peptides bore this label. These data indicate that the fluorescent rTTR derivatives are relatively uniform with respect to molecular mass and site of fluorophore substitution. Therefore, straightforward interpretation of dissociation experiments is warranted within the constraints indicated herein.
Sedimentation velocity experiments were conducted on the fluorescent rTTR conjugates to examine structural modulation by intrinsic (cysteine adducts) and extrinsic (diflunisal) factors. In preliminary analyses conducted at pH 7, no dissociation of the tetramer for any of the three species was observed. However, the experiments were limited in sensitivity due to the low quantum yield of fluorescein at neutral pH. Concentrations below ∼14 nm tetramer demonstrated typical S/N of <15 dB, rendering analysis problematic. To enable the measurements at significantly lower concentrations and to improve the S/N, the preliminary experiments were duplicated in buffer of pH 9, where FITC fluorescence is maximal. Under these conditions, the S/N level of 15 dB was not reached until the concentration was reduced to ∼0.2 nm tetramer. Thus, measurement in alkaline buffer allowed an ∼70-fold increase in detector sensitivity.
In order to determine how alkaline pH affects TTR structure, near and far UV circular dichroism spectra for F-rTTR and rTTR in pH 7 and pH 9 buffers were generated (Fig. 1). In far UV experiments (Fig. 1A), the spectra for rTTR at pH 7 (solid line) and F-rTTR at pH 9 (dashed line) overlap, indicating that neither the covalently bound fluorescein moiety nor increased alkalinity significantly alter rTTR secondary structure. Furthermore, the spectra in the near UV range (Fig. 1B) for rTTR at pH 7 (solid line) and pH 9 (dashed line) exhibited only minor differences in amplitude, primarily in the spectral range of <270 nm, suggesting that rTTR tertiary structure is largely conserved between pH 7 and 9. Sedimentation velocity of F-rTTR in pH 9 buffer at relatively high concentrations indicated a single ideal species with a sedimentation coefficient of 4.320 S (Fig. 2), as expected for tetrameric TTR. This result was identical to the corresponding experiment in pH 7 buffer (data not shown), suggesting that the hydrodynamic volume of solvated F-rTTR tetramer at the two pH values is similar and further supporting the conservation of structure at alkaline pH.
FIGURE 1.

Alkaline pH does not alter native TTR secondary or tertiary structure. CD spectra for 12 μm solutions of rTTR and F-rTTR in 10 mm Tris, 100 mm KCl, pH 7 and pH 9, were collected at 25 °C. A, far UV spectra for rTTR at pH 7 (solid line) and F-rTTR at pH 9 (dashed line). B, near UV spectra for rTTR at pH 7 (solid line) and pH 9(dashed line).
FIGURE 2.

Example of c(s) analysis of F-rTTR sedimentation velocity. F-rTTR (110 nm tetramer in 0.1 mg/ml ovalbumin, 100 mm potassium chloride, 10 mm Tris, pH 9) was sedimented at 50,000 rpm with a rotor temperature of 20 °C. Data were acquired with the fluorescence optical system and analyzed with the c(s) fitting model in the program SEDFIT. A, raw data (○) and finite element fit (lines) with time- and position-invariant signal offsets removed. For clarity of presentation, only every fifth scan of the data set is shown. B, best fit residuals of the presented scans. C, c(s) distribution representing the 68% confidence interval of the fit. The sedimentation coefficient determined from the peak of the distribution is indicated.
The fluorescent rTTR derivatives were largely homogeneous with respect to monomer mass and site of conjugation, allowing for straightforward interpretation of the s̄ isotherms, assuming that the bound dye molecule itself was not a major contributor to quaternary structural stability. To test this possibility, a 2-fold dilution series of F-rTTR as well as 1:1 F-rTTR/rTTR over the tetramer concentration range of 0.43–110 nm were analyzed by sedimentation velocity. Comparison of the resulting s̄ isotherms (not shown) indicated a slight shift to lower concentrations in the isotherm of the mixed tetrameric species. Furthermore, modeling of the isotherms with the program SEDPHAT indicated a decrease of ∼1.6-fold in the c½ value of the mixture relative to the homogeneous species. Together, these data suggest that the FITC adduct is slightly destabilizing and that the presented interpretations of the s̄ isotherms will render conservative estimates of the energy of self-association. However, it was reasoned that this bias will not affect comparisons among the three F-rTTR derivatives.
The concentration dependence of s̄ for F-rTTR, F-rTTR-SO3H, and F-rTTR-Cys was investigated. Although this type of analysis is model-independent (28), considerable information about the molecular composition of reacting species can be obtained by evaluation of the c(s) distributions at each concentration, an analytical approach that assumes discrete noninteracting species. As indicated in Fig. 3, relatively high concentrations of F-rTTR-Cys (109 nm tetramer) yielded a single peak at ∼4.3 S. At lower concentrations, two peaks were apparent, with the lower indicating a sedimentation coefficient of ∼1.7 S. The peak corresponding to the larger sedimentation coefficient broadened and decreased considerably with decreasing concentration, whereas the peak position of the smaller sedimentation coefficient was relatively unchanged with concentration. These results are qualitatively consistent with Gilbert-Jenkins theory and support the use of c(s) analysis for extracting qualitative information from reversibly associating systems (37). In no case did s values greater than ∼4.3 S appear at dissociating concentrations, suggesting that if aggregation of monomeric subunits was occurring, it was not observable in our analyses.
FIGURE 3.

The family of c(s) curves for F-rTTR-Cys indicates tetramer dissociation. Various concentrations of F-rTTR-Cys were subjected to sedimentation velocity at 50,000 rpm with constant rotor temperature of 20 °C. Data were acquired with the fluorescence optical system and analyzed with the program SEDFIT. Distributions for the indicated loading concentrations were generated with the c(s) fitting model.
Buffer component-corrected s̄20, W isotherms were generated for F-rTTR, F-rTTR-SO3H, and F-rTTR-Cys (Fig. 4). In all three cases, s̄20, W decreased with decreasing concentration, a hallmark of self-associating systems. At high concentrations, the isotherms for F-rTTR and F-rTTR-SO3H both plateau at ∼4.6 S, consistent with the expectation for tetrameric TTR (∼56 kDa). F-rTTR tetramer dissociation was detected initially as low as 6.90 nm tetramer, whereas F-rTTR-SO3H did not dissociate significantly until a much lower concentration (0.43 nm tetramer). Conversely, dissociation of F-rTTR-Cys was detected at the highest concentration measured (109 nm tetramer).
FIGURE 4.

Cys10 adducts modulate the quaternary structure stability of F-rTTR. Concentration-dependent s̄20, W isotherms for F-rTTR (○), F-rTTR-SO3H(□), and F-rTTR-Cys (▵) were generated by sedimentation velocity analyses. All samples were diluted in 0.1 mg/ml ovalbumin, 100 mm potassium chloride, 10 mm Tris, pH 9, and experiments were conducted with fluorescence detection at 50,000 rpm with constant rotor temperature of 20 °C. Data points and error intervals for each species were generated with the Monte Carlo for weight-average s values simulation routine using the best fit parameters determined by c(s) analyses. Apparent s̄ values were converted to s̄20, W values with the program SEDNTERP and modeled with the program SEDPHAT. The resulting best fit dissociation curves are indicated for each species (F-rTTR(4→1) (solid line), F-rTTR-SO3H(4→1) (dashed line), and F-rTTR-Cys(4→2→1) (dotted line)).
It should be noted that, in no case were s̄20, W values consistent with monomeric TTR (∼1.8 S) detected, and therefore complete isotherms could not be obtained. Concentrations lower than those indicated in Fig. 4 were measured; however, the S/N of the data at these concentrations were determined to be too low to allow rigorous analyses. Although there is no established cut-off for acceptable S/N in sedimentation velocity analysis, it was determined that at S/N of <5 dB, small changes in fitting parameters (i.e. meniscus position) led to large changes in the Monte Carlo simulation result without an appreciable change to the r.m.s.d. of the finite-element fit of the data, rendering interpretation problematic.
The shape of the isotherms could be modeled using the program SEDPHAT, despite the lack of reliable data at concentrations below ∼0.2 nm tetramer. Models for 1→4 and 1→2→4 association were applied to the data from each species and assessed by the F-statistic. For F-rTTR, the fits to 1→4 and 1→2→4 models were not statistically different (F = 2.27). For F-rTTR-SO3H, no stable fit for the 1→2→4 reaction was attained. Thus, the data for these two species were most appropriately interpreted in the context of 1→4 reactions. For F-rTTR-Cys, the fit to the 1→2→4 model was significantly better (F = 4.89). The fitting results and corresponding r.m.s.d. values are indicated in Table 1. For all three species, low c½ or KD values were determined. The S-sulfonated form was determined to be the most stable (c½ = 32.9 pm), followed by the unmodified form (c½ = 237.6 pm), and S-cysteinylated form (KD4→2 = 532.5 pm). Although these values are expected to be conservative estimates of binding strength because of the destabilizing presence of FITC, assessment of the relative contributions of the modifications is probably unaffected. Thus, it is estimated that S-sulfonation stabilized the TTR tetramer by ∼7-fold, whereas S-cysteinylation destabilized it by ∼2-fold with respect to the c½ of the unmodified form.
TABLE 1.
Modeling of the s̄20, W isotherms for F-rTTR Cys-10 adducts
| Species | r.m.s.d. 1→4 | c½ | r.m.s.d. 1→2→4 | KD4→2 | KD2→1 |
|---|---|---|---|---|---|
| pm | pm | pm | |||
| F-rTTR | 0.0626 | 237.6 | 0.0415 | 172.3 | 585.3 |
| F-rTTR-SO3Ha | 0.0555 | 32.9 | |||
| F-rTTR-Cys | 0.1538 | 839.0 | 0.0696 | 532.5 | 879.5 |
A stable fit to a 1→2→4 reaction model was not observed.
In addition to examining the effect of cysteine adduction on TTR tetramer stability, the concentration dependence of F-rTTR s̄ was also determined in the presence and absence of diflunisal. As indicated in Fig. 5, at higher concentrations of F-rTTR, both in the absence and presence of diflunisal, a plateau of s̄ at values consistent with TTR tetramer (∼4.3 S) was observed. In the absence of diflunisal, the F-rTTR s̄ isotherm decreased with diminishing concentration in the range below 6.90 nm tetramer. In the presence of diflunisal, no decrease in s̄ was detected, and only a single peak was observed between 1 and 5 S in the c(s) distributions of each concentration tested.
FIGURE 5.

Diflunisal inhibits F-rTTR tetramer dissociation. Sedimentation velocity was used to generate s̄ isotherms for F-rTTR in the absence (○) and presence (□) of diflunisal. Data sets were analyzed with the program SEDFIT using a combination of c(s) analysis to determine the best fit parameters and the Monte Carlo for weight-average s values simulation routine to determine s̄ with its associated error space. Lines are included to aid visual interpretation and do not represent regression analysis of the data.
The described experiments were conducted in dilute buffers, and the data could be modeled as thermodynamically and hydrodynamically ideal solutions of the Lamm equation (36). As proof of concept of fluorescence-detected sedimentation velocity in complex, concentrated biological fluids, F-rTTR was diluted to 6.25 μg/ml (110 nm tetramer) in human serum and centrifuged at 50,000 rpm. The data profile with finite element fit, the residuals of the fit, and the resulting c(s) distribution are shown in Fig. 6. A single, sharp sedimenting boundary was observed (Fig. 6A), moving at an apparent sedimentation coefficient of 2.4 S (Fig. 6C). Although there is a slight slope to the plateau and some residual signal above the boundary at lower radial positions, these features were not fit by the Lamm equation, as evidenced by the lack of additional peaks in the c(s) distribution. There is no evidence that the boundary is distorted by the Johnston-Ogston effect (40, 41). Despite the qualitative agreement between the sedimentation coefficient determined by Lamm equation modeling and that determined by boundary midpoint migration (Fig. 6D), the systematic residuals (Fig. 6B) show that the shape of the boundary could not be recapitulated by this model. Although better fits to the data were obtained by increasing the fitting parameter corresponding to the frictional ratio (f/f0), the meaning of f/f0 in complex, concentrated solutions like serum is uncertain.
FIGURE 6.

Example of c(s) analysis of F-rTTR sedimentation in serum. F-rTTR was diluted in human serum to 110 nm tetramer and subjected to sedimentation velocity at 50,000 rpm, 20 °C. Data were acquired with the fluorescence optical system and analyzed with the c(s) fitting model in the program SEDFIT. A, raw data (○) and finite element fit (lines) with time- and position-invariant signal offsets removed. For clarity, only every fifth scan of the data set is shown. The half-height of the plateau signal (determined from the first scan immediately adjacent to the data roll-off) is indicated with a horizontal dashed line. B, best fit residuals of the presented scans. C, c(s) distribution representing the 68% confidence interval of the fit. The sedimentation coefficient determined from the peak of the distribution is indicated. D, qualitative determination of s indicated by the slope of a plot of ln r/rm versus ω2t. The linear regression of the data, with a slope of 2.452 × 10–13 s (2.452 S), is indicated below along with the correlation coefficient (r2) of the regression.
DISCUSSION
Aggregation of TTR is characteristic of two distinct forms of systemic amyloidosis. Most of the models proposed to describe this process involve dissociation of the native tetramer as the initial and rate-limiting step. Therefore, factors that promote structural instability are potential initiators of disease onset. Conversely, factors that stabilize the native structure would inhibit aggregation, thereby protecting against disease onset. We hypothesized that Cys10 adducts could affect TTR quaternary structure and modulate the rate-limiting step of wild-type TTR fibrillogenesis.
We have established fluorescence-detected analytical ultracentrifugation as a viable tool in the study of the relationship between TTR chemical structure and the energy of self-association. This primary analytical method allows direct insight into such systems by the determination of the concentration dependence of s̄. Due to the very high stability of the native wild-type TTR tetramer, such analyses became possible only with the recent development of fluorescence optics for the analytical ultracentrifuge (30), which are several orders of magnitude more sensitive than the existing commercial optical systems.
For our studies, rTTR was produced and modified at Cys10 by S-sulfonation and S-cysteinylation to generate the two most predominant adducts in human serum (17–20). These species, as well as unmodified rTTR, were covalently bound to the amine-reactive fluorescein derivative, FITC. The concentration dependence of the weight-average sedimentation coefficient was then determined for each form.
One requirement for interpreting structural stability data from proteins labeled with a fluorescent moiety is that the effect of the dye on the overall structure must be characterized. In systems where dye molecules can conjugate to multiple sites on the protein, the possibility of structural destabilization increases with higher dye/monomer stoichiometries. In this study, the effect of FITC conjugation on the stability of the rTTR tetramer was established in two ways. First, the sample heterogeneity with respect to mass and derivatization site was thoroughly characterized by ESI-MS and LC-MS/MS of a tryptic digest. By ESI-MS, the preparations were found to be largely homogeneous with respect to mass (>85%), with the predominant product having 1:1 dye/monomer stoichiometry. We further confirmed by LC-MS/MS that this product was labeled at the N-terminal Gly residue and not in detectable quantities at any of the internal Lys residues. These observations suggested that the destabilizing effects of the dye would be minimal in our preparation. Second, in addition to characterizing the heterogeneity and site specificity, we directly examined the effect of fluorescein labeling on tetramer stability by comparing the s̄ isotherm of F-rTTR with that of a 1:1 mixture of labeled/unlabeled rTTR. The estimated 1.6-fold decrease in c½ between the mixture and pure F-rTTR indicated only slight destabilization by the dye, confirming our expectation of minimal structural perturbation while suggesting that our determinations of c½ would be conservative.
Determination of s̄ as a function of concentration by sedimentation velocity is a classical approach to the analysis of self-associating systems (26–29). In our study, the determination of s̄ was made for each data set in a two-stage process using the sedimentation analysis program SEDFIT. In the first stage, c(s) distribution analysis was used to determine the best fit of the data. These analyses are model-dependent and treat sedimenting boundaries as discrete, noninteracting species. However, in addition to providing best fit parameters for subsequent model-independent analyses, evaluation of the c(s) distribution at each concentration (see Fig. 3) gives several insights into the thermodynamic behavior of the system.
This approach is similar in practice to the method of evaluating the family of g(s) curves, well described in studies of tubulin polymerization (see Ref. 26 and references therein). First, self-associating systems of finite stoichiometry will approximate discrete, noninteracting species at concentrations where either association is highly favored (high concentrations) or dissociation is highly favored (low concentrations) by mass action. Under these conditions, c(s) distribution analysis provides a reliable estimate of s as well as the diffusion coefficient (D) and therefore the molecular weight of the species. When the molecular weight of the monomer is known, by mass spectrometry for example, this analysis then provides the stoichiometry of the species in solution. In the analyses of F-rTTR species, the model-dependent c(s) analysis performed at high concentrations (∼110 nm tetramer) in all three cases yielded a species consistent with an ∼56-kDa tetramer (∼4.3 S). Therefore, subsequent model-independent s̄ analyses were interpreted within the context of tetramer dissociation. A second insight obtained in the analysis of the c(s) family of curves for self-associating systems is that an estimate of s̄ is obtained by integration across the distribution. At intermediate concentrations, the data cannot be modeled as discrete, noninteracting species. However, in these cases, c(s) peaks represent the reaction boundaries of the association (37). Therefore, the determination of s̄ by integration, although not analytically rigorous, is valid qualitatively and provides independent evidence to support subsequent model-independent analyses.
The second stage of analysis in this study was the quantitative determination of s̄ by the Monte Carlo simulation routine in SEDFIT. This analysis provides a rigorous, model-independent determination of s̄ and its associated error space, assuming that the best fit parameters have been assigned. An isotherm can then be constructed by converting the s̄ values to s̄20, W values and then plotting as a function of loading concentration. By this approach, the isotherms for F-rTTR, F-rTTR-SO3H, and F-rTTR-Cys were constructed and compared. There is a clear difference in the concentration dependence of s̄20, W among the three species (Fig. 4). Tetramers composed of F-rTTR-SO3H began to dissociate at a 16-fold lower concentration than did F-rTTR tetramers, suggesting a stabilizing effect for S-sulfonation. Conversely, at the highest concentration measured, the s̄20, W of F-rTTR-Cys was slightly lower than expected for the tetramer, suggesting that detectable dissociation was already occurring. This concentration was almost 16-fold higher than the concentration where F-rTTR dissociation was initially detected, suggesting that S-cysteinylation destabilized the quaternary structure of rTTR.
These interpretations were supported and quantified by modeling of the s̄20, W isotherms with the program SEDPHAT. Models for 1→4 association were more appropriate for the F-rTTR and F-rTTR-SO3H data, whereas the F-rTTR-Cys isotherm was fit significantly better with the 1→2→4 model. That different models were appropriate for modified forms of the same protein is not surprising, since 1→4 association is a special case of 1→2→4 association where the dimer state is not appreciably populated (KD2→1→∞). Our results suggest that S-cysteinylation affects the stability of both the tetramer and dimer but to different extents. This is not surprising, considering that the intermolecular interactions comprising the interfaces between monomeric subunits are not identical (the tetramer is composed of a dimer of dimers). Despite these differences in model appropriateness, the three F-rTTR species can be compared with respect to initial (tetramer) dissociation values (c½ for F-rTTR and F-rTTR-SO3H and KD4→2 for F-rTTR-Cys). Our results indicate an ∼7-fold decrease in the c½ of the S-sulfonated form relative to the unmodified form. Conversely, the KD4→2 of the S-cysteinylated form was increased by ∼2-fold over the c½ of the unmodified form. It should be noted that, in all three cases, the fits demonstrated only moderate r.m.s.d. values. This is probably due to the fact that these analyses assume linearity in detector response between monomeric and tetrameric species. It is possible that association induces collisional quenching of FITC, leading to nonlinearity of the fluorescence signal with respect to association state. This effect cannot be accurately measured or controlled at this point. However, it is expected that, due to the relatively steep transitions in the concentration dependence of s̄20, W, this effect will result in only minor errors in the determination of dissociation constants.
In recent years, interest has arisen in exploring the potential of therapeutic strategies that interfere with amyloid formation by stabilizing the native state of the precursor protein (42–45). TTR aggregation has been a model system for this approach, because small molecules, including the nonsteroidal anti-inflammatory drug diflunisal and many of its derivatives, are reported to stabilize the native tetrameric structure (31, 32, 46). In this study, we directly investigated the effect of diflunisal on F-rTTR tetramer structure by analysis of the s̄ isotherms in the presence and absence of drug. It is clear in this analysis that diflunisal completely eliminated measurable F-rTTR dissociation in the examined concentration range. This is the first report of direct analysis of TTR native state stabilization and suggests that fluorescence-detected analytical ultracentrifugation may serve as a useful platform for screening potential drug candidates that inhibit TTR tetramer dissociation.
The analysis of F-rTTR quaternary structure modulation by cysteine adducts and diflunisal reported herein was obtained by sedimentation velocity, a primary analytical method. As such, the measurements are governed solely by thermodynamic principles and are completely independent of solvent conditions. Thus, the effects of potential structural modulators can be determined in solvents favoring native amino acid packing arrangements. Therefore, the shape of the s̄ isotherm is dictated by quaternary structural rearrangements and is independent of tertiary or secondary structure. By this logic, our results suggest that Cys10 adduction, as well as diflunisal, modulate TTR stability by altering native contacts between the subunits. As noted, ovalbumin was included as a carrier protein in our experiments. In addition, an alkaline, nonphysiologic pH was used for all samples to maximize the fluorescence properties of FITC. In general, these two factors deviate from standard biophysical practice. However, it should be noted that these are practical limitations based on experimental considerations and are not indicative of theoretical shortcomings. In addition, our results are in qualitative accord with complementary analyses that have suggested a stabilizing effect of S-sulfonation and diflunisal and a destabilizing effect of S-cysteinylation on wild-type TTR structure under denaturing conditions (31, 47).
The analyses of the F-rTTR s̄ isotherms presented herein were possible because the high sensitivity of the fluorescence optical system (30) allowed monitoring of the dissociation of the tightly interacting TTR subunits. Furthermore, the selectivity of the optical system enabled detection of F-rTTR against a high background of nonfluorescent co-solutes. Sedimentation studies in such complex solutions have previously been accomplished only with preparative centrifugation followed by fractionation and detection of tracer molecules by offline fluorescence, enzymatic, or immunologic assays (48). Here, we demonstrated the use of fluorescence-detected sedimentation velocity to analyze F-rTTR in a complex, concentrated biological fluid (i.e. serum). This result has tremendous importance because the use of first principles methods to investigate effectors of TTR quaternary structure in such mixtures allows data to be interpreted in the context of the native, physiologic milieu. As Fig. 6 makes clear, the most pressing limitation to this type of analysis is not experimental but lies in the analytical treatment of the data. Rigorous treatment of sedimentation data by finite element fitting of the sedimentation transport equation (36, 49) or by time-derivative analysis (50, 51) requires that the behavior of the sedimenting species be governed by well defined parameters. Sedimentation in heterogeneous, complex mixtures is affected by a number of contributing factors arising from the sedimentation of high concentrations of co-solutes. These include the unmixing of co-solute complexes, the formation of concentration gradients, and the presence of variable solution density and viscosity. These complicating factors are not yet characterized well enough to incorporate into rigorous data analysis.
Despite the lack of a detailed model for sedimentation in biological fluids, our analysis suggests that a qualitative treatment of the data may be applied, at least in the case of TTR, thereby facilitating the understanding of these contributions. It is clear from Fig. 6A that the finite element fit of the transport equation intersects with the actual data near the approximate boundary midpoint, suggesting that the sedimentation coefficient obtained from the c(s) distribution (2.4 S) approximates the actual sedimentation coefficient of F-rTTR at 6.25 μg/ml in serum. In addition, this value is in qualitative agreement with the sedimentation coefficient determined by boundary midpoint migration (Fig. 6D), a method that simplifies the determination of s by ignoring the effects of diffusion and solute-solute interactions on boundary shape. Our studies suggest that it may be possible to investigate TTR self-association using c(s) distribution analyses in a physiologically relevant environment, such as serum. In this context, the measurement of the concentration dependence of the sedimentation coefficient may provide an improved, more realistic model of self-association. It is not yet clear whether this approach would be applicable to systems other than TTR; however, the ∼2 S decrease in s when determined in serum as opposed to dilute buffer is consistent with observations for other similarly sized proteins (41).
The present study, for the first time, demonstrates the quaternary structural modulation of TTR by both intrinsic (cysteine adducts) and extrinsic (diflunisal) effectors from thermodynamic first principles. The results described herein support the hypothesis that Cys10 adducts modulate the onset of SSA (or ATTR) by altering the population of monomeric (aggregation-prone) with respect to tetrameric (stable) TTR molecules. Specifically, S-cysteinylation may promote disease onset, whereas S-sulfonation may protect against it. It is anticipated that fluorescence-detected analytical ultracentrifugation will provide a useful experimental paradigm not only for further studies of TTR structure but also in the investigation of other tightly associating systems similar to TTR.
Acknowledgments
We thank Giuseppe Infusini for assistance with the LTQ-Orbitrap hybrid mass spectrometer.
This work was supported by American Heart Association Grant AHA0060149T (to L. H. C.), National Institutes of Health Grants P41 RR10888 and S10 RR10493 (to C. E. C.) and S10 RR020946 (to J. Zaia), the Gerry Foundation, the Young Family Amyloid Research Fund, the Eileen Cochran Amyloid Research Fund, and the David S. Levine Amyloid Research Fund. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TTR, transthyretin; ATTR, familial TTR-related amyloidosis; SSA, senile systemic amyloidosis; FITC, fluorescein-5-isothiocyanate; rTTR, recombinant human transthyretin; rTTR-SO3H, S-sulfonated rTTR; rTTR-Cys, S-cysteinylated rTTR; ESI, electrospray ionization; MS, mass spectrometry; LC, liquid chromatography; HPLC, high pressure liquid chromatography; F-rTTR, FITC-labeled rTTR; r.m.s.d., root mean square deviation.
References
- 1.Walley, V. M., Kisilevsky, R., and Young, I. D. (1995) Cardiovasc. Pathol. 4 79–102 [DOI] [PubMed] [Google Scholar]
- 2.Falk, R. H., and Skinner, M. (2000) Adv. Intern. Med. 45 107–137 [PubMed] [Google Scholar]
- 3.Merlini, G., and Bellotti, V. (2003) N. Engl. J. Med. 349 583–596 [DOI] [PubMed] [Google Scholar]
- 4.Connors, L. H., Lim, A., Prokaeva, T., Roskens, V. A., and Costello, C. E. (2003) Amyloid 10 160–184 [DOI] [PubMed] [Google Scholar]
- 5.Benson, M. D., and Uemichi, T. (1996) Amyloid 3 44–56 [Google Scholar]
- 6.Westermark, P., Sletten, K., Johansson, B., and Cornwell, G. G., III (1990) Proc. Natl. Acad. Sci. U. S. A. 87 2843–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright, J. R., and Calkins, E. (1975) J. Am. Geriatr. Soc. 23 97–103 [DOI] [PubMed] [Google Scholar]
- 8.Hodkinson, H. M., and Pomerance, A. (1977) Q. J. Med. 46 381–387 [PubMed] [Google Scholar]
- 9.Cornwell, G. G., III, Murdoch, W. L., Kyle, R. A., Westermark, P., and Pitkänen, P. (1983) Am. J. Med. 75 618–623 [DOI] [PubMed] [Google Scholar]
- 10.Smith, T. J., Kyle, R. A., and Lie, J. T. (1984) Mayo Clin. Proc. 59 547–555 [DOI] [PubMed] [Google Scholar]
- 11.Ng, B., Connors, L. H., Davidoff, R., Skinner, M., and Falk, R. H. (2005) Arch. Intern. Med. 165 1425–1429 [DOI] [PubMed] [Google Scholar]
- 12.Kelly, J. W. (1998) Curr. Opin. Struct. Biol. 8 101–106 [DOI] [PubMed] [Google Scholar]
- 13.Damas, A. M., and Saraiva, M. J. (2000) J. Struct. Biol. 130 290–299 [DOI] [PubMed] [Google Scholar]
- 14.Jiang, X., Smith, C. S., Petrassi, H. M., Hammarström, P., White, J. T., Sacchettini, J. C., and Kelly, J. W. (2001) Biochemistry 40 11442–11452 [DOI] [PubMed] [Google Scholar]
- 15.Quintas, A., Vaz, D. C., Cardoso, I., Saraiva, M. J., and Brito, R. M. (2001) J. Biol. Chem. 276 27207–27213 [DOI] [PubMed] [Google Scholar]
- 16.Hurshman, A. R., White, J. T., Powers, E. T., and Kelly, J. W. (2004) Biochemistry 43 7365–7381 [DOI] [PubMed] [Google Scholar]
- 17.Théberge, R., Connors, L. H., Skinner, M., Skare, J., and Costello, C. E. (1999) Anal. Chem. 71 452–459 [DOI] [PubMed] [Google Scholar]
- 18.Suhr, O. B., Svendsen, I. H., Ohlsson, P.-I., Lendoire, J., Trigo, P., Tashima, K., Ranløv, P. J., and Ando, Y. (1999) Amyloid 6 187–191 [DOI] [PubMed] [Google Scholar]
- 19.Lim, A., Prokaeva, T., McComb, M. E., O'Connor, P. B., Théberge, R., Connors, L. H., Skinner, M., and Costello, C. E. (2002) Anal. Chem. 74 741–751 [DOI] [PubMed] [Google Scholar]
- 20.Lim, A., Sengupta, S., McComb, M. E., Théberge, R., Wilson, W. G., Costello, C. E., and Jacobsen, D. W. (2003) J. Biol. Chem. 278 49707–49713 [DOI] [PubMed] [Google Scholar]
- 21.Lashuel, H. A., Wurth, C., Woo, L., and Kelly, J. W. (1999) Biochemistry 38 13560–13573 [DOI] [PubMed] [Google Scholar]
- 22.Hammarström, P., Sekijima, Y., White, J. T., Wiseman, R. L., Lim, A., Costello, C. E., Altland, K., Garzuly, F., Budka, H., and Kelly, J. W. (2003) Biochemistry 42 6656–6663 [DOI] [PubMed] [Google Scholar]
- 23.Svedberg, T., and Pedersen, K. O. (1940) The Ultracentrifuge, pp. 28–29, Oxford, London
- 24.Schachman, H. K. (1959) Ultracentrifugation in Biochemistry, pp. 157–170, Academic Press, Inc., New York
- 25.Cann, J. R. (1970) Interacting Macromolecules: The Theory and Practice of Their Electrophoresis, Ultracentrifugation and Chromatography, pp. 133–149, Academic Press, Inc., New York
- 26.Correia, J. J. (2000) Methods Enzymol. 321 81–100 [DOI] [PubMed] [Google Scholar]
- 27.Stafford, W. F. (2000) Methods Enzymol. 323 302–325 [DOI] [PubMed] [Google Scholar]
- 28.Schuck, P. (2003) Anal. Biochem. 320 104–124 [DOI] [PubMed] [Google Scholar]
- 29.Sontag, C. A., Stafford W. F., and Correia, J. J. (2004) Biophys. Chem. 108 215–230 [DOI] [PubMed] [Google Scholar]
- 30.MacGregor, I. K., Anderson, A. L., and Laue, T. M. (2004) Biophys. Chem. 108 165–185 [DOI] [PubMed] [Google Scholar]
- 31.Miller, S. R., Sekijima, Y., and Kelly, J. W. (2004) Lab. Invest. 84 545–552 [DOI] [PubMed] [Google Scholar]
- 32.Adamski-Werner, S. L., Palaninathan, S. K., Sacchettini, J. C., and Kelly, J. W. (2004) J. Med. Chem. 47 355–374 [DOI] [PubMed] [Google Scholar]
- 33.Kingsbury, J. S., Klimtchuk, E. S., Théberge, R., Costello, C. E., and Connors, L. H. (2007) Protein Expression Purif. 53 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raz, A., and Goodman, D. S. (1969) J. Biol. Chem. 244 3230–3237 [PubMed] [Google Scholar]
- 35.Mann, M., Meng, C. K., and Fenn, J. B. (1989) Anal. Chem. 61 1702–1708 [Google Scholar]
- 36.Schuck, P. (2000) Biophys. J. 78 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dam, J., and Schuck, P. (2005) Biophys. J. 89 651–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnaible, V., and Pryzbylski, M. (1999) Bioconjugate Chem. 10 861–866 [DOI] [PubMed] [Google Scholar]
- 39.Teske, C. A., Simon, R., Niebisch, A., and Hubbuch, J. (2007) Biotechnol. Bioeng. 98 193–200 [DOI] [PubMed] [Google Scholar]
- 40.Johnston, J. P., and Ogston, A. G. (1946) Trans. Faraday Soc. 42 789–799 [Google Scholar]
- 41.Kroe, R. A. (2005) Applications of Fluorescence Detected Sedimentation, Ph.D. Thesis, University of New Hampshire, Durham, NH
- 42.Miroy, G. J., Lai, Z., Lashuel, H. A., Peterson, S. A., Strang, C., and Kelly, J. W. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 15051–15056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiti, F., Taddei, N., Stefani, M., Dobson, C. M., and Ramponi, G. (2001) Protein Sci. 10 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason, J. M., Kokkoni, N., Stott, K., and Doig, A. J. (2003) Curr. Opin. Struct. Biol. 13 526–532 [DOI] [PubMed] [Google Scholar]
- 45.Ray, S. S., Nowak, R. J., Brown, R. H., Jr., and Lansbury, P. T., Jr. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almeida, M. R., Macedo, B., Cardoso, I., Alves, I. L., Valencia, G., Arsequell, G., Planas, A., and Saraiva, M. J. (2004) Biochem. J. 381 351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Q., and Kelly, J. W. (2003) Biochemistry 42 8756–8761 [DOI] [PubMed] [Google Scholar]
- 48.Rivas, G., Fernandez, J. A., and Minton, A. P. (1999) Biochemistry 38 9379–9388 [DOI] [PubMed] [Google Scholar]
- 49.Demeler, B., and Saber, H. (1998) Biophys. J. 74 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stafford, W. F., III (1992) Anal. Biochem. 203 295–301 [DOI] [PubMed] [Google Scholar]
- 51.Philo, J. S. (2000) Anal. Biochem. 279 151–163 [DOI] [PubMed] [Google Scholar]