

Abstract
Pathogenic Yersinia spp. carry a large common plasmid that encodes a number of essential virulence determinants. Included in these factors are the Yersinia-secreted proteins called Yops. We analyzed the consequences of wild-type and mutant strains of Yersinia pseudotuberculosis interactions with the macrophage cell line RAW264.7 and murine bone marrow-derived macrophages. Wild-type Y. pseudotuberculosis kills ≈70% of infected RAW264.7 macrophages and marrow-derived macrophages after an 8-h infection. We show that the cell death mediated by Y. pseudotuberculosis is apoptosis. Mutant Y. pseudotuberculosis that do not make any Yop proteins no longer cause host cell death. Attachment to host cells via invasin or YadA is necessary for the cell death phenotype. Several Yop mutant strains that fail to express one or more Yop proteins were engineered and then characterized for their ability to cause host cell death. A mutant with a polar insertion in YpkA Ser/Thr kinase that does not express YpkA or YopJ is no longer able to cause apoptosis. In contrast, a mutant no longer making YopE or YopH (a tyrosine phosphatase) induces apoptosis in macrophages similar to wild type. When yopJ is added in trans to the ypkAyopJ mutant, the ability of this strain to signal programmed cell death in macrophages is restored. Thus, YopJ is necessary for inducing apoptosis. The ability of Y. pseudotuberculosis to promote apoptosis of macrophages in cell culture suggests that this process is important for the establishment of infection in the host and for evasion of the host immune response.
Keywords: type III secretion, programmed cell death, AvrRxv, Yersinia-secreted protein
Yersinia species cause a variety of diseases. Yersinia pestis is the causative agent of plague, and Yersinia pseudotuberculosis and Yersinia enterocolitica cause adenitis, septicemia, and gastrointestinal syndromes. Although the route of infection differs for Y. pestis compared with Y. pseudotuberculosis and Y. enterocolitica, they share a common tropism for lymphoid tissues (1).
The enteropathogenic Yersinia make four products that are important for attachment and penetration of the intestinal barrier. Three of these products are encoded on the chromosome (invasin, Ail, and Psa) (2–4) and one (YadA) is plasmid-encoded (5). YadA and invasin bind mammalian cells expressing β1 integrins (6, 7). In addition to YadA, there are a number of other genetic factors encoded on the plasmid pYV that are essential for bacterial pathogenicity. Many of these plasmid genes are known to be part of a host cell contact-dependent or type III secretory pathway (1). The coordinate activities of the secretion machinery and the adherence factors allows the bacteria to introduce or translocate other plasmid proteins (Yops) directly into the target cell cytoplasm (1).
Macrophages and polymorphonuclear leukocytes become nonphagocytic after infection by wild-type Yersinia (1). The invading bacteria form extracellular microcolonies on the surface of the inert phagocytes (8–10). Several groups have shown that two Yop proteins, YopE and YopH, are responsible for this inhibition of phagocytosis (11, 12). YopE causes actin depolymerization and rounding up of host cells by an unknown mechanism (13). YopH is a tyrosine phosphatase (14) that can inhibit the Fc receptor-mediated oxidative burst in macrophages (15). Both YopE and YopH are essential for virulence. YpkA, another Yersinia-secreted protein, is a Ser/Thr protein kinase and also has been shown to be essential for virulence (16). YopJ is another secreted protein that is encoded by a gene that is cotranscribed with ypkA and has no known biochemical function (17).
Recently a number of studies have shown that apoptosis is triggered in host cells in response to infection by a variety of extra- and intracellular bacterial pathogens (18–21). Apoptosis is caused by a variety of mechanisms including inhibition of host cell protein synthesis by bacterial A-B toxins (22), disruption of membrane integrity by pore-forming proteins or hemolysins [Staphylococcus aureus α-toxin (23) or enteropathogenic Escherichia coli hemolysin (24)], and activation of the caspase interleukin 1β converting enzyme by IpaB from Shigella (25).
In this study, we demonstrate that RAW264.7 and murine bone marrow-derived macrophages (BMM) infected with Y. pseudotuberculosis or Y. enterocolitica show clear manifestations of apoptosis. YopE and YopH are not involved in the signaling of macrophage apoptosis. Y. pseudotuberculosis that is deficient for the full-length YpkA protein product and YopJ no longer signaled programmed cell death in infected macrophages. The ability to kill macrophages is restored by expressing YopJ on a multi-copy plasmid.
MATERIALS AND METHODS
Bacterial Strains and Growth Conditions.
Yersinia strains and sources are as follows: YPIIIpIB1, wild-type Y. pseudotuberculosis (26); YPIIIpIB71, lcr (5); YPIIIpIB102, yadA (5); YPIIIpYVIII6, yopE (7); YP66pIB1, inv (7); YP66pIB102, inv yadA (7). YPIIIp506, ypkA; YPIIIp507, ypkAyopE; YPIIIp502, yopH; YPIIIp503, yopEyopH (details of the construction of these strains will be published elsewhere; D.M.M., J.M., Bärbel Raupach, and S.F., unpublished data); 8081c, wild-type Y. enterocolitica (26); SL1344, wild-type Salmonella typhimurium (20).
Yersinia strains were grown overnight in 2× YT medium at 26°C. The day of the assay the bacteria were diluted 1:50 into 2× YT plus 20 mM sodium oxalate and 20 mM MgCl2 and grown with aeration for 2 h at 26°C and then shifted to 37°C and grown with aeration for 2 h. S. typhimurium were grown as described (20). E. coli strains were grown in Luria broth or on Luria agar.
Cell Death Assays in 96-Well Plates.
Twenty thousand RAW264.7 (ATCC TIB71) cells or BMM [isolated as described (27)] were seeded into 96-well plates 15–18 h before use. Monolayers were infected with a moi of 50:1 and centrifuged at 165 × g for 5 min to synchronize the infection. Following a 2-h incubation at 37°C (5% CO2), gentamicin was added to a final concentration of 50 μg/ml. Supernatants of the infected macrophages were sampled and assayed for lactate dehydrogenase (LDH) using the cytotox 96 kit (Promega) according to the manufacturer’s instructions. The percent cell death was calculated as 100 × (experimental release − spontaneous release)/(total release − spontaneous release), where spontaneous release is the amount of LDH activity in supernatants of cells incubated in medium alone, and total release is the activity from macrophages lysed with 1% Triton X-100. Yersinia strains do not have endogenous LDH activity when grown aerobically.
Assessment of Apoptosis by Terminal Deoxynucleotidyltransferase-Mediated dUTP Nick End Labeling (TUNEL) Reaction and Electron Microscopy.
Macrophages infected with Y. pseudotuberculosis were analyzed for the presence of DNA fragmentation using the TUNEL reaction. The In Situ Cell Death Detection Kit for Fluorescein (Boehringer Mannheim) was used to label free 3′-OH termini of DNA fragments with fluorescein. Infections on coverslips were performed as described (20). The fixed macrophages were incubated with polyclonal rabbit anti-Y. pseudotuberculosis antiserum, followed by goat anti-rabbit 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-conjugated antibody (Vector Laboratories), stained with rhodamine phalloidin (Molecular Probes), and analyzed by fluorescence microscopy. Infected samples for electron microscopy were prepared exactly as described (28).
Analysis of Secreted Yops.
Yersinia strains were grown as described for cell death assays. The secreted proteins were precipitated with 10% trichloroacetic acid and subjected to SDS/PAGE. The proteins were transferred to nitrocellulose by standard Western blotting procedures (29). The blot was then incubated in rabbit anti-YopJ polyclonal antisera (generous gift from Olaf Shneewind, University of California at Los Angeles). The secondary antibody was anti-rabbit horseradish peroxidase conjugate and the detection system was the ECL chemilluminescence kit (Amersham).
Cloning of ypkA and yopJ.
Plasmid DNA isolations, restriction enzyme digestions, ligations, and transformations of E. coli were performed essentially as described (30). Standard PCRs were used to clone ypkA and yopJ. Oligonucleotide primers flanking the ORF1 and ypkA coding regions were used to clone ypkA into the SalI and SphI sites of pACYC184 (31): 5′-GAGTGAGCATGCGATTTGGCAATTGCTTAACAA-3′ and 5′-GTGTGATCTAGACAGATAACGGCAGCGAAT-3′ from yopJ sequence were used to clone into the SphI and XbaI sites of pACYC184.
RESULTS
Yersinia Kill Macrophages in Vitro.
Both Y. pseudotuberculosis and Y. pestis are known to kill macrophages in vitro and it has previously been shown that the observed cytotoxicity is dependent on some virulence plasmid encoded factor (32). In this study, we compared the abilities of wild-type Y. pseudotuberculosis with various Y. pseudotuberculosis mutants deficient in either adherence properties or various Yops to kill macrophages. We used a cell death assay that measures the release of a macrophage cytoplasmic enzyme, LDH, into the tissue culture medium. Wild-type Y. pseudotuberculosis killed 25–30% of infected macrophages after 4 h and up to 75% of the phagocytes by 8 h postinfection (Table 1, row a). However, a mutant strain, YP66 pIB102 (inv−, YadA−), that is competent in Yop secretion but is unable to adhere to cells did not kill macrophages (Table 1, row b). Mutants expressing either invasin, YPIIIpIB102, or YadA, YP66, killed macrophages to a similar level as wild type. The YadA+ invasin− strain killed slightly higher numbers, 66.3 ± 3.2%, of macrophages than the YadA− invasin+ strain, 40.6 ± 6.9% (P = 0.0402) at 6 h (Table 1, rows c and d). A Y. pseudotuberculosis mutant in lcr, which does not produce any of the secreted proteins, was unable to kill macrophages (Table 1, row e). Because bacteria that adhered to macrophages but were Yop deficient did not induce host cell death, we conclude that either invasin or YadA were needed for adherence and the Yop delivery was essential for Yersinia-induced macrophage death.
Table 1.
YopJ and invasin or YadA are necessary for macrophage death
| row | Strain | % cell death*
|
|||
|---|---|---|---|---|---|
| 2 h | 4 h | 6 h | 8 h | ||
| a | YPIII pIB1 | 2.62 ± 2.84 | 27.41 ± 4.36 | 38.77 ± 2.23 | 73.93 ± 0.46 |
| b | YP66 pIB102 | 0.00 ± 0.52 | 0.00 ± 0.92 | 0.00 ± 0.00 | ND |
| c | YPIII pIB102 | 0.00 ± 0.73 | 13.36 ± 1.82 | 40.56 ± 4.90 | ND |
| d | YP66 pIB1 | 0.00 ± 1.71 | 25.09 ± 1.27 | 66.3 ± 3.18 | ND |
| e | YPIII pIB71 | 0.00 ± 0.42 | 0.08 ± 1.21 | 0.00 ± 2.19 | 3.05 ± 3.05 |
| f | YPIII pYVIII6 | 0.00 ± 0.78 | 54.28 ± 2.77 | 55.32 ± 2.70 | 66.9 ± 1.84 |
| g | YPIII p502 | 0.32 ± 0.48 | 47.27 ± 4.23 | 48.47 ± 4.33 | 68.42 ± 7.71 |
| h | YPIII p503 | 0.00 ± 1.21 | 46.73 ± 6.28 | 47.93 ± 6.14 | 60.95 ± 3.04 |
| i | YPIII p506 | 0.00 ± 0.31 | 0.00 ± 0.58 | 0.00 ± 0.56 | 3.26 ± 3.88 |
| YPIII p506(pypkA) | 0.00 ± 0.48 | 0.25 ± 1.2 | 0.00 ± 0.00 | 2.23 ± 1.54 | |
| j | YPIII p506(pyopJ) | 0.00 ± 0.48 | 18.21 ± 4.06 | 19.98 ± 3.97 | 40.58 ± 5.79 |
| YPIII p506(pACYC184) | 0.00 ± 1.55 | 0.94 ± 3.12 | 0.27 ± 0.53 | 6.76 ± 3.61 | |
| YPIII p507 | 0.00 ± 0.23 | 0.06 ± 3.77 | 2.19 ± 3.66 | 4.05 ± 3.58 | |
| YPIII p507(pypkA) | 0.52 ± 0.89 | 0.00 ± 1.21 | 0.00 ± 0.35 | 2.34 ± 1.82 | |
| k | YPIII p507(pyopJ) | 0.00 ± 2.30 | 22.91 ± 8.81 | 31.66 ± 2.39 | 60.72 ± 7.80 |
| YPIII p507(pACYC184) | 0.00 ± 1.43 | 0.00 ± 0.87 | 3.80 ± 3.55 | 5.13 ± 3.79 | |
| l | 8081c | 0.00 ± 0.00 | 59.68 ± 3.83 | 54.29 ± 7.90 | 60.84 ± 0.88 |
| m | SL1344 | 89.05 ± 10.38 | 97.41 ± 13.71 | 100 ± 5.55 | ND |
| SL1344† | 33.47 ± 5.16 | 45.60 ± 8.78 | 63.82 ± 2.11 | ND | |
ND, not determined.
Mean percentage of macrophage death ± standard deviation from a representative Cytotox96 LDH release assay with moi of 50 bacteria per RAW264.7 cell.
SL1344, S. typhimurium, moi of 10 bacteria per macrophage.
We also compared Yersinia-induced macrophage death with Salmonella-induced macrophage death (20). Two hours postinfection, nearly 90% of the macrophages infected with wild-type, invasive S. typhimurium at a moi of 50:1 were dead, whereas the percentage of dead macrophages infected with wild-type Y. pseudotuberculosis at a similar moi remained at background (Table 1, rows m and a). This difference implies that S. typhimurium and Y. pseudotuberculosis mechanisms of macrophage cell death might be different.
To determine which of the known Yops were involved in the induction of macrophage death, we analyzed various strains defective in the production of one or more Yops. A strain that no longer produces YopE, a molecule responsible for host cell actin depolymerization, still killed macrophages to a similar degree as wild type (Table 1, row f). Likewise, a strain deficient for production of YopH, a tyrosine pyrophosphatase, as well as a strain unable to produce both YopE and YopH were able to kill macrophages with similar kinetics and to the same level as the wild-type Y. pseudotuberculosis and Y. enterocolitica (Table 1, rows g, h, and l). However, a mutant deficient for production of YpkA and YopJ (due to a polar insertion in ypkA) did not kill macrophages (Table 1, row i). Thus, neither YopE nor YopH were involved in causing macrophage death, but either or both YpkA and YopJ were necessary for Yersinia-induced death.
Yersinia Induces Apoptosis in Macrophages.
We examined RAW264.7 cells infected with wild-type bacteria and various mutant derivatives by transmission electron microscopy. Within 6 h postinfection, wild-type Y. pseudotuberculosis were found tightly adherent to the surface of most of the macrophages or seem to be “embedded” in the plasma membrane. Many infected macrophages displayed intense perinuclear chromatin aggregation, cytoplasmic vacuolization, and maintenance of organelle structure, all characteristic of cells undergoing apoptosis (Fig. 1) (33). Likewise, macrophages infected with bacteria unable to make YopE or YopH displayed the same characteristics of apoptosis (Fig. 1). In contrast, macrophages exposed to bacteria unable to make YpkA or YopJ showed a normal morphology, similar to the cells infected with the Y. pseudotuberculosis Yop− strain (Fig. 1).
Figure 1.

Transmission electron micrographs of apoptotic RAW264.7 macrophages infected with Y. pseudotuberculosis. Macrophages were infected with YPIIIpIB1 wild-type Y. pseudotuberculosis, YPIIIp506 (ypkAyopJ mutant), YPIIIp503 (yopEyopH mutant), or YPIIIpIB71 Yop mutant for 6 h. Note the chromatin condensation, cytoplasmic vacuolization, and swollen endoplasmic reticulum in macrophages infected with YPIIIpIB1 and YPIIIp503. In contrast, the macrophages infected with YPIIIp506 or YPIIIpIB71 appear normal despite the presence of bacteria.
We also examined infected cells by scanning electron microscopy to analyze the effects of Y. pseudotuberculosis on the macrophage membrane. Within 4 h postinfection, many of the host cells exposed to wild-type Y. pseudotuberculosis as well as those infected with the double yopEyopH mutant bacteria displayed dramatic membrane blebbing, characteristic of apoptosis (Fig. 2) (34). In contrast, the macrophages infected with the ypkAyopJ mutant did not display the membrane blebbing, although some cell rounding and disappearance of filopodia was apparent compared with uninfected RAW264.7 macrophages (Fig. 2).
Figure 2.

Scanning electron micrographs of infected RAW264.7 macrophages. Macrophages were infected for 6 h with (a) wild-type Y. pseudotuberculosis YPIIIpIB1, (b) YPIIIp506, (c) YPIIIp503, or (d) uninfected control. Note membrane blebbing on the surface of cells infected with YPIIIpIB1 and YPIIIp503. Macrophages infected with YPIIIp506 appear slightly rounded compared to uninfected cells, but the membrane blebbing is absent.
Another characteristic of apoptosis is the cleavage of DNA of the dying cell at the internucleosomal regions, resulting in multimers of 180–200 bp (35, 36). We used the TUNEL reaction (37) to detect DNA fragmentation in BMM infected with various strains of Y. pseudotuberculosis and Y. enterocolitica. By 6 h postinfection with wild-type Y. pseudotuberculosis or with the yopEyopH double mutant, nearly all of the BMM were undergoing apoptosis (Fig. 3). Similar results were obtained with Y. enterocolitica-infected BMM (data not shown). Macrophages infected with the ypkAyopJ double mutant strain of Y. pseudotuberculosis or the Y. pseudotuberculosis strain lacking secretion of all Yops exhibited no more apoptosis than uninfected macrophages. Thus, the ypkAyopJ mutant could not induce apoptosis in primary macrophages.
Figure 3.

Infections with (a) YPIIIpIB1, (b) YPIIIp506, (c) YPIIIp503, or (d) YPIIIpIB71 for 8 h. TUNEL reaction was used to label 3′-OH termini with fluorescein. Anti-Y. pseudotuberculosis primary antibody and anti-rabbit 7-amino-4-methylcoumarin-3-acetic acid (AMCA) secondary antibody were used to label bacteria and rhodamine phalloidin was used to label actin filaments. Images of epifluorescence were scanned into Adobe photoshop and aligned to make a composite. The arrow indicates an infected macrophage positive for TUNEL reaction.
Complementation of Polar ypkA Mutant with yopJ.
To determine whether YpkA and/or YopJ were required for apoptosis, we cloned the ypkA structural gene and its promoter into the vector, pACYC184, which replicates in Y. pseudotuberculosis. When the resulting plasmid was expressed in the ypkAyopJ mutant strain, it did not complement the polar ypkA mutation on the virulence plasmid even though we detected the 82-kDa YpkA protein on a Coomassie-stained SDS/PAGE of secreted bacterial proteins (data not shown). When the yopJ gene was added in trans (by cloning it into pACYC184) to the ypkAyopJ and ypkAyopJyopE mutant strains, the ability to kill macrophages was restored (Table 1, rows j and k). The production of the YopJ product was confirmed by demonstrating the presence of a 32.5-kDa protein on a Coomassie-stained SDS/PAGE which bound a YopJ-specific rabbit polyclonal antisera in a Western blot (Fig. 4).
Figure 4.
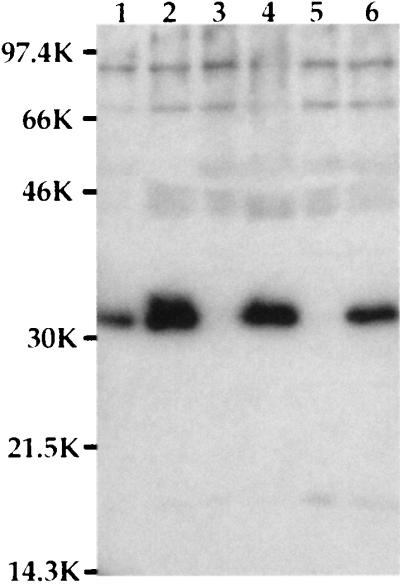
YopJ expression. Supernatants from bacteria grown as described were precipitated with trichloroacetic acid and run on SDS/PAGE. The proteins were transferred to nitrocellulose, and the 32.5-kDa YopJ protein was detected with anti-YopJ antibody. Lanes: 1, YPIIpIB1; 2, YPIIIp503; 3, YPIIIp506; 4, YPIIIp506(pyopJ); 5, YPIIIp506(pACYC184); 6, YPIIIp507(pyopJ).
We performed TUNEL reactions on macrophages infected with the ypkAyopJ mutant strain expressing YopJ in trans to determine if the macrophage death observed in the LDH cell death assay was indeed apoptosis. The number of cells displaying positive TUNEL reactions and the density of the monolayer at 6 h postinfection with the ypkAyopJ mutant strain containing YopJ was very similar to results obtained with wild-type Y. pseudotuberculosis, whereas cells infected with the ypkAyopJ mutant containing pACYC184 gave no more apoptosis than uninfected controls (data not shown).
DISCUSSION
We have shown that Y. pseudotuberculosis kills macrophages by triggering a pathway for programmed cell death. For this to occur, the bacteria need to attach intimately to the macrophage membrane. When neither of two bacterial adhesins, invasin and YadA, are expressed, Y. pseudotuberculosis does not kill macrophages despite the production and secretion of the Yops. This tight attachment to β1 integrins has been shown to be critical for the bacteria to efficiently deliver effector molecules (Yops) to the host cell via a type III secretion system (7, 13). We also observed that a strain containing a mutation in a gene required for expression of all Yops, lcr, no longer killed macrophages and concluded that at least one of the Yops was necessary for triggering apoptosis.
To determine which Yop(s) mediated apoptosis, we tested a variety of mutant Y. pseudotuberculosis strains defective for Yop production. The ypkA insertion mutant we tested did not produce either YpkA or YopJ and no longer mediated apoptosis in macrophages. The ability of this mutant to kill macrophages by apoptosis was restored when yopJ alone was expressed from a recombinant plasmid, not when ypkA was expressed. Thus, YopJ is required for Yersinia-induced apoptosis, however, YopJ alone may not be sufficient. The ypkAyopJ mutant strain retained the ability to translocate the effector molecules, YopE and YopH, into the macrophage as demonstrated by the antiphagocytic phenotype displayed in the transmission electron microscopy and scanning electron microscopy (Figs. 1 and 2) and the depolymerized actin within infected macrophages (rhodamine phalloidin staining; Fig. 3). Thus, YopJ is not required for the translocation of these known Yersinia effector proteins.
The blastp (38) program was used to compare YopJ to the current protein databases (Protein Indentification Resource, Swiss-Protein, and Genpept). One protein, AvrRxv (42.1 kDa), an avirulence protein from a bacterial plant pathogen, Xanthomonas campestris pv. vesicatoria (39), had significant homology to four regions of YopJ. The smallest sum probability score for the alignment of these two proteins was 4 × 10−5. All avr gene expression results in host cell death in the infected plant cells (40). The exact mechanism by which AvrRxv interacts with host cells is not known. Although, several avr genes have been found to be important for pathogen fitness and/or symptom formation (40). Another X. campestris pv. vesicatoria avr protein, AvrBs3, has recently been reported to be localized inside the pepper plant cell (41).
We have shown that YopE and YopH, which are necessary for the antiphagocytic phenotype of Y. pseudotuberculosis, are not involved in inducing apoptosis. Although we and others have reported that YopE and YopH are responsible for causing monolayers of epithelial and fibroblast cells to lift off in tissue culture, the type of cell death was not characterized (7, 26). The macrophage death described here is probably a different mechanism from what has been described in epithelial and fibroblast cell lines. In fact, in these cell lines we see no apoptotic cell death (unpublished data). Further studies are underway to determine the mechanism of epithelial and fibroblast cell death(s).
Questions still remain as to whether YopJ is the actual effector molecule that interacts directly with host cell proteins and is sufficient for signaling macrophage apoptosis. A possible role of YopJ in Yersinia-induced macrophage death is that YopJ directly activates the host cell’s programmed cell death machinery either upstream of the activation of caspases or by directly activating a caspase in the same way that IpaB from Shigella binds and activates interleukin 1β converting enzyme (25). Because Salmonella makes a protein highly homologous to IpaB, SipB, which is necessary for invasion of host cells (42) and for inducing apoptosis in macrophages (ref. 19; unpublished results), it is probably activating apoptosis in macrophages by a mechanism very similar to Shigella (our data and personal communication by A. Zychlinsky, Skirball Institute, New York University School of Medicine). The result shown here that S. typhimurium kills macrophages very rapidly compared with Y. pseudotuberculosis suggests that Y. pseudotuberculosis may signal apoptosis by activating a programmed cell death pathway at a different step than Shigella and S. typhimurium.
How might the ability of Y. pseudotuberculosis to induce macrophage apoptosis in vitro contribute to in vivo virulence? Y. pseudotuberculosis remains mainly extracellular in the host; thus, it would be advantageous for Y. pseudotuberculosis to neutralize one of the host’s primary defenses, macrophages, before they have the opportunity to produce proinflammatory cytokines such as interleukin 6 and tumor necrosis factor α. Although the YopJ protein has been reported to be dispensable for virulence (17), the ability to induce apoptosis in macrophages could aid in the establishment of infection in the host and for evasion of the host immune system.
Although significant progress has been made in identifying molecules involved in apoptosis, mechanisms and regulation of this essential pathway are still largely unsolved. Thus, elucidating the bacterial molecules and the mechanisms by which Y. pseudotuberculosis and other bacterial pathogens trigger apoptosis of host cells may be valuable in dissecting host cell pathways of programmed cell death. The homology between YopJ and AvrRxv leads one to speculate that cell death mechanisms in plant and animal cells may be conserved.
Acknowledgments
We thank Dr. Alex Hromockyj for supplying antibodies recognizing Yersinia, Dr. Olaf Schneewind (University of California, Los Angeles) for generously supplying YopJ antiserum, Dr. David P. Discher and Dr. Arturo Zychlinsky for critical reading of the manuscript. We also thank Dr. Guy Cornelis for pointing out the X. campestris AvrRxv and YopJ protein homologies. This work was supported by National Institutes of Health Grant AI26195 and Defense Advanced Research Planning Agency contract MDA972-97-K-0001. J.M. was supported by Damon Runyon–Walter Winchell Cancer Research Fund Grant 1277.
ABBREVIATIONS
- BMM
bone marrow-derived macrophage
- LDH
lactate dehydrogenase
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
References
- 1.Cornelis G R, Wolf-Watz H. Mol Microbiol. 1997;23:861–867. doi: 10.1046/j.1365-2958.1997.2731623.x. [DOI] [PubMed] [Google Scholar]
- 2.Isberg R R, Falkow S. Nature (London) 1985;317:262–264. doi: 10.1038/317262a0. [DOI] [PubMed] [Google Scholar]
- 3.Miller V L, Falkow S. Infect Immun. 1988;56:1242–1248. doi: 10.1128/iai.56.5.1242-1248.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Merriam J J, Mueller J P, Isberg R R. Infect Immun. 1996;64:2483–2489. doi: 10.1128/iai.64.7.2483-2489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolin I, Wolf-Watz H. Infect Immun. 1984;43:72–78. doi: 10.1128/iai.43.1.72-78.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isberg R R, Leong J M. Cell. 1990;60:861–871. doi: 10.1016/0092-8674(90)90099-z. [DOI] [PubMed] [Google Scholar]
- 7.Bliska J B, Copass M C, Falkow S. Infect Immun. 1993;61:3914–3921. doi: 10.1128/iai.61.9.3914-3921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanski C, Naumann M, Grutzkau A, Pluschke G, Friedrich B, Hahn H, Riecken E O. Infect Immun. 1991;59:1106–1111. doi: 10.1128/iai.59.3.1106-1111.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heesemann J, Hantke K, Vocke T, Saken E, Rakin A, Stojiljkovic I, Berner R. Mol Microbiol. 1993;8:397–408. doi: 10.1111/j.1365-2958.1993.tb01583.x. [DOI] [PubMed] [Google Scholar]
- 10.Simonet M, Richard S, Berche P. Infect Immun. 1990;58:841–845. doi: 10.1128/iai.58.3.841-845.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fallman M, Andersson K, Hakansson S, Magnusson K E, Stendahl O, Wolf-Watz H. Infect Immun. 1995;63:3117–3124. doi: 10.1128/iai.63.8.3117-3124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosqvist R, Bolin I, Wolf-Watz H. Infect Immun. 1988;56:2139–2143. doi: 10.1128/iai.56.8.2139-2143.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosqvist R, Forsberg A, Rimpilainen M, Bergman T, Wolf-Watz H. Mol Microbiol. 1990;4:657–667. doi: 10.1111/j.1365-2958.1990.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 14.Bliska J B, Guan K L, Dixon J E, Falkow S. Proc Natl Acad Sci USA. 1991;88:1187–1191. doi: 10.1073/pnas.88.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bliska J B, Black D S. Infect Immun. 1995;63:681–685. doi: 10.1128/iai.63.2.681-685.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galyov E E, Hakansson S, Forsberg A, Wolf-Watz H. Nature (London) 1993;361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- 17.Galyov E E, Hakansson S, Wolf-Watz H. J Bacteriol. 1994;176:4543–4548. doi: 10.1128/jb.176.15.4543-4548.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzman C A, Domann E, Rohde M, Bruder D, Darji A, Weiss S, Wehland J, Chakraborty T, Timmis K N. Mol Microbiol. 1996;20:119–126. doi: 10.1111/j.1365-2958.1996.tb02494.x. [DOI] [PubMed] [Google Scholar]
- 19.Chen L M, Kaniga K, Galan J E. Mol Microbiol. 1996;21:1101–1115. doi: 10.1046/j.1365-2958.1996.471410.x. [DOI] [PubMed] [Google Scholar]
- 20.Monack D M, Raupach B, Hromockyj A E, Falkow S. Proc Natl Acad Sci USA. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Zychlinsky A. Microbiol Pathog. 1994;17:203–212. doi: 10.1006/mpat.1994.1066. [DOI] [PubMed] [Google Scholar]
- 22.Kochi S K, Collier R J. Exp Cell Res. 1993;208:296–302. doi: 10.1006/excr.1993.1249. [DOI] [PubMed] [Google Scholar]
- 23.Jonas D, Walev I, Berger T, Liebetrau M, Palmer M, Bhakdi S. Infect Immun. 1994;62:1304–1312. doi: 10.1128/iai.62.4.1304-1312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonas D, Schultheis B, Klas C, Krammer P H, Bhakdi S. Infect Immun. 1993;61:1715–1721. doi: 10.1128/iai.61.5.1715-1721.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Smith M R, Thirumalai K, Zychlinsky A. EMBO J. 1996;15:3853–3860. [PMC free article] [PubMed] [Google Scholar]
- 26.Portnoy D A, Moseley S L, Falkow S. Infect Immun. 1981;31:775–782. doi: 10.1128/iai.31.2.775-782.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren M K, Vogel S N. J Immunol. 1985;134:982–989. [PubMed] [Google Scholar]
- 28.Pascopella L, Raupach B, Ghori N, Monack D, Falkow S, Small P L. Infect Immun. 1995;63:4329–4335. doi: 10.1128/iai.63.11.4329-4335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 30.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Rose R E. Nucleic Acids Res. 1988;16:355. doi: 10.1093/nar/16.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goguen J D, Walker W S, Hatch T P, Yother J. Infect Immun. 1986;51:788–794. doi: 10.1128/iai.51.3.788-794.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerr J F R, Wyllie A H, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arends M J, Wyllie A H. Int Rev Exp Pathol. 1991;32:223–254. doi: 10.1016/b978-0-12-364932-4.50010-1. [DOI] [PubMed] [Google Scholar]
- 35.Wyllie A H. Nature (London) 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 36.Arends M J, Morris R G, Wyllie A H. Am J Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- 37.Gavrieli Y, Sherman Y, Ben-Sasson S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Whalen M C, Wang J F, Carland F M, Heiskell M E, Dahlbeck D, Minsavage G V, Jones J B, Scott J W, Stall R E, Staskawicz B J. Mol Plant–Microbe Interact. 1993;6:616–627. doi: 10.1094/mpmi-6-616. [DOI] [PubMed] [Google Scholar]
- 40.Dangl J L. Curr Top Microbiol Immunol. 1994;192:99–118. doi: 10.1007/978-3-642-78624-2_5. [DOI] [PubMed] [Google Scholar]
- 41.Van den Ackerveken G, Marois E, Bonas U. Cell. 1996;87:1307–1316. doi: 10.1016/s0092-8674(00)81825-5. [DOI] [PubMed] [Google Scholar]
- 42.Hermant D, Menard R, Arricau N, Parsot C, Popoff M Y. Mol Microbiol. 1995;17:781–789. doi: 10.1111/j.1365-2958.1995.mmi_17040781.x. [DOI] [PubMed] [Google Scholar]