

An atomic resolution structure of the copper-binding domain of the Alzheimer’s disease amyloid precursor protein is presented.
Keywords: Alzheimer’s disease, amyloid precursor protein, copper protein, ultrahigh resolution
Abstract
Amyloid precursor protein (APP) plays a central role in the pathogenesis of Alzheimer’s disease, as its cleavage generates the Aβ peptide that is toxic to cells. APP is able to bind Cu2+ and reduce it to Cu+ through its copper-binding domain (CuBD). The interaction between Cu2+ and APP leads to a decrease in Aβ production and to alleviation of the symptoms of the disease in mouse models. Structural studies of CuBD have been undertaken in order to better understand the mechanism behind the process. Here, the crystal structure of CuBD in the metal-free form determined to ultrahigh resolution (0.85 Å) is reported. The structure shows that the copper-binding residues of CuBD are rather rigid but that Met170, which is thought to be the electron source for Cu2+ reduction, adopts two different side-chain conformations. These observations shed light on the copper-binding and redox mechanisms of CuBD. The structure of CuBD at atomic resolution provides an accurate framework for structure-based design of molecules that will deplete Aβ production.
1. Introduction
Alzheimer’s disease is characterized by extensive neuronal death and the formation of abnormal protein aggregates that compromise normal brain functions such as memory and cognition. Although the cause of the disease has not been pinpointed, there is considerable evidence that a peptide called Aβ contributes to its pathogenesis (Selkoe, 2002 ▶). This peptide has a tendency to aggregate into small soluble oligomers, eventually leading to the amyloid aggregates observed in diseased brains. In particular, small soluble oligomers of Aβ have been shown to cause damage to cultured neurons (Klyubin et al., 2005 ▶; Lesné et al., 2006 ▶; Haass & Selkoe, 2007 ▶). Therefore, a potential approach to therapeutics for Alzheimer’s disease is to decrease the production of Aβ. The Aβ peptide arises from the cleavage of amyloid precursor protein (APP), a type I transmembrane protein with a large extracellular portion containing several structural and functional domains (Kang et al., 1987 ▶). APP is cleaved in one of two principal pathways. In the amyloidogenic pathway, APP is first cleaved at a site predicted to be near the membrane by the β-site APP-cleaving enzyme (Sinha et al., 1999 ▶; Yan et al., 1999 ▶), followed by an intramembranous cleavage by the γ-secretase complex (Iwatsubo, 2004 ▶) to generate the Aβ peptide. Alternatively, APP may initially be cleaved within the Aβ sequence by the α-secretase enzyme (Esch et al., 1990 ▶); the resultant peptide is not capable of forming aggregates.
The putative functions of APP include neuronal growth and guidance (Rossjohn et al., 1999 ▶), signalling (Nishimoto et al., 1993 ▶; Okamoto et al., 1995 ▶) and copper metabolism (Maynard et al., 2005 ▶). This last function may be attributed to the extracellular copper-binding domain (CuBD). Importantly for the pathogenesis of Alzheimer’s disease, the interaction between Cu2+ ions and CuBD appears to decrease APP cleavage through the amyloidogenic pathway (Borchardt et al., 1999 ▶) and mutation of the copper-binding residues oblates the effect (Borchardt et al., 2000 ▶). The beneficial effects of Cu2+ have been demonstrated in transgenic mice studies, in which elevation of Cu2+ levels by feeding or overexpression of a transmembrane copper transporter improved the survival of the mice, with a concomitant decrease in Aβ levels in the brain (Bayer et al., 2003 ▶; Phinney et al., 2003 ▶). However, Cu2+ will not be suitable for therapeutics, since human CuBD is capable of reducing Cu2+ to Cu+ (Multhaup et al., 1996 ▶), which is neurotoxic (White, Multhaup et al., 1999 ▶).
We have previously determined the structures of CuBD in the apo form and with Cu ions bound (Barnham et al., 2003 ▶; Kong et al., 2007 ▶). The binding of Cu caused some rather subtle changes to the Cu-binding pocket through small rotations of the side chains of His147, His151 and Tyr168, which were identified as the copper-binding residues. The Cu-coordination geometry changed from distorted square pyramidal in the +2 state to distorted square planar in the +1 state through the loss of an apical water. Apart from these differences, the changes in the geometric parameters around the active site were again subtle. We now report the structure of the apo form of CuBD determined to ultrahigh resolution, which enables a more detailed analysis of the protein. High-resolution structures typically allow a more comprehensive description of protein motion and function. A higher resolution data set contains a greater amount of data, so that more parameters can be introduced into the model. Normal restraints on amino-acid geometry need not be imposed, so that individual atomic coordinates can be refined free of bias towards idealized values (Schmidt & Lamzin, 2002 ▶). Atoms of similar mass, such as carbon, nitrogen and oxygen, are often distinguishable in the electron density, allowing accurate positioning of histidine, glutamine and asparagine side chains, and in some cases H atoms may become evident (Schmidt & Lamzin, 2002 ▶). The mobility of each atom can be modelled anisotropically by six directional parameters instead of a single B factor (Schneider, 1996 ▶). Together with better visualization of alternative side-chain conformations of lower occupancy, the overall protein mobility can thus be better discerned. High resolution is also beneficial for elucidating enzyme-reaction mechanisms and studying interactions with ligands (Vrielink & Sampson, 2003 ▶) because of the more detailed structural information. The high-resolution structure reported here reveals structural features that further our understanding of the Cu-binding function of APP.
2. Materials and methods
2.1. Structure determination and refinement
The CuBD crystals used in this work were grown and cryoprotected as described elsewhere (Kong et al., 2007 ▶). Diffraction data were collected on beamline 14-BM-C of the Advanced Photon Source synchrotron (Chicago, USA) from a crystal frozen at 100 K. The wavelength of the beamline was fixed at 0.9 Å and data were collected on an ADSC Quantum-315 detector. Two rounds of data collection were performed. Firstly, a high-resolution pass was used to record higher resolution diffraction, which is more sensitive to radiation damage. A longer exposure time (3 s) was employed to improve the signal from the weaker high-resolution data and 180° of data were collected. A low-resolution pass followed, using a shorter exposure time (1 s) so that the diffraction spots would not become overloaded. The high- and low-resolution passes were indexed separately by the HKL program DENZO and then integrated together using SCALEPACK (Otwinowski & Minor, 1997 ▶). The data-collection statistics are presented in Table 1 ▶.
Table 1. Data collection and processing.
Values in parentheses are for the highest resolution shell (0.87–0.85 Å).
| Temperature (K) | 100 |
| Wavelength (Å) | 0.9 |
| Space group | P212121 |
| Unit-cell parameters (Å) | |
| a | 31.3 |
| b | 32.5 |
| c | 50.1 |
| Maximum resolution (Å) | 0.85 |
| No. of crystals | 1 |
| No. of observations | 245165 |
| No. of unique reflections | 41418 |
| Data completeness (%) | 90.5 (43.8) |
| I/σ(I) | 23.2 (1.7) |
| Multiplicity | 5.9 (1.5) |
| Rmerge† (%) | 6.0 (37.0) |
R
merge = 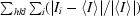 , where I
i is the intensity of the ith measurement of an equivalent reflection with indices hkl and 〈I〉 is the mean of these measurements.
, where I
i is the intensity of the ith measurement of an equivalent reflection with indices hkl and 〈I〉 is the mean of these measurements.
The reflection file from SCALEPACK was first converted to CNS format (Brünger et al., 1998 ▶) for phasing by rigid-body refinement (resolution range 20–2.0 Å) using the previous apo CuBD structure (PDB code 2fjz; Kong et al., 2007 ▶) with solvent molecules omitted. Instead of using the original test set (10% of all reflections), a new set of 5% was picked because it was desired to include more data in the calculation of the structure. Potential model bias was overcome by simulated annealing at 3000 K (using data in the resolution range 20–0.9 Å) after rigid-body refinement. Further refinement was carried out in the SHELX-97 suite of programs (Sheldrick, 1997 ▶), starting with restrained conjugate-gradient least-squares refinement (CGLS) and followed by extension to the maximal resolution of 0.85 Å and the use of full anisotropic refinement. An examination of the 2F obs − F calc electron-density map at this point (contoured at the 1.5σ level) permitted the building of alternate side-chain conformations for Met170 and Glu183. Water molecules were added to the model and one glycerol molecule could be identified clearly at a distance of about 4 Å from the His147 side chain. The geometry restraints of glycerol were generated from the PRODRG website (Schüttelkopf & van Aalten, 2004 ▶). Geometric restraints were relaxed gradually except for the side chains of Glu131, Ala132, His151 and Ile176, because some of the bond geometries in those residues were found to deviate up to 9 standard deviations from the Engh and Huber values (Engh & Huber, 1991 ▶). H atoms were then added to all atoms of the protein according to the riding hydrogen model. The diffuse solvent parameter SWAT was refined to allow a better bulk-solvent correction. Following further rounds of CGLS refinement, R work and R free of the model reached 13.1% and 15.0%, respectively. The final refinement statistics are presented in Table 2 ▶.
Table 2. Refinement statistics.
| Non-H atoms | |
| Protein | 492 |
| Glycerol | 6 |
| Solvent (H2O) | 115 |
| Resolution (Å) | 0.85 |
| Rwork† (%) | 13.1 |
| Rfree†‡ (%) | 15.0 |
| Reflections used in Rwork calculations | |
| No. of reflections | 2107 |
| Completeness (%) | 86.0 |
| R.m.s.d.s from ideal geometry | |
| Bonds (Å) | 0.03 |
| Angles (°) | 2.4 |
| Dihedrals (°) | 24.9 |
| Impropers (°) | 1.7 |
| Bonded B (Å2) | |
| Main chain | 1.3 |
| Side chain | 2.2 |
| Mean isotropic B (protein) (Å2) | 10.3 |
| Main chain | 9.0 |
| Side chain | 11.6 |
| Mean isotropic B (solvent) (Å2) | 25.3 |
| Mean isotropic B (glycerol) (Å2) | 14.1 |
| Residues in Ramachandran plot (%) | |
| Most favoured regions | 90.4 |
| Additional allowed regions | 9.6 |
R = 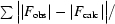
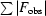 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated with 5% of the diffraction data that were selected randomly and not used throughout refinement.
2.2. Structure analysis
The geometry of the final model was checked with PROCHECK (Laskowski et al., 1993 ▶) and the Biotech Validation Suite for Protein Structures (http://biotech.ebi.ac.uk:8400). The anisotropy of the structure was analysed using the SHELXPRO program in the SHELX package and the online Protein Anisotropic Refinement Validation and Analysis Tool (PARVATI; Merritt, 1999 ▶).
3. Results and discussion
3.1. Structure determination
The structure of CuBD was determined to 0.85 Å resolution, with final R and R free values of 13.1% and 15.0%, respectively, which are within the range observed for sub-angstrom resolution structures (Kleywegt & Jones, 2002 ▶). Various analyses indicated a high-quality structure; for example, 90.4% of dihedral angles were found to be in the most favourable region of the Ramachandran plot. The bond lengths and angles of the final model are within 5 standard deviations of ideal values (Engh & Huber, 1991 ▶). (Note that four residues required restraints; see §3.4.)
The structure contains residues 133–189 of the native protein. All three disulfide bonds remained intact despite the use of synchrotron radiation and their dihedral angles were analysed (Table 3 ▶) and classified according to established criteria (Richardson, 1981 ▶). The Cys133–Cys187 pair belongs to a short right-handed hook which can be found between adjacent antiparallel β-strands in other proteins and is sometimes called a ‘staple’ (Harrison & Sternberg, 1994 ▶). The Cys158–Cys186 pair is typical of a left-handed spiral, which is quite common in proteins. The Cys144–Cys174 pair can be classified as an ‘unusually long disulfide’ in which the χ2 angles of the cysteines are close to 180° and the Cα atoms are separated by a longer distance than in other disulfide conformations. This disulfide bond appears to be strained, as a χ2 range of 60–120° is more energetically favourable (Richardson, 1981 ▶).
Table 3. A list of dihedral angles and Cα—Cα distances in the three disulfides of the high-resolution CuBD structure.
The χ1′ and χ2′ values refer to the second cysteine, while χ3 refers to the dihedral angle of the disulfide bond.
| Firstcysteine | Secondcysteine | χ1 (°) | χ2 (°) | χ3 (°) | χ2′ (°) | χ1′ (°) | Cα—Cα (Å) |
|---|---|---|---|---|---|---|---|
| Cys133 | Cys187 | −53 | −113 | 103 | −74 | −67 | 4.12 |
| Cys144 | Cys174 | 53 | 180 | 81 | −165 | −60 | 6.23 |
| Cys158 | Cys186 | −67 | −48 | −79 | −60 | −58 | 5.34 |
3.2. Structure comparison
The crystal used for this 0.85 Å atomic resolution structure (Fig. 1 ▶) is of the same crystal form as the previous 1.6 Å resolution apo structure reported by Kong et al. (2007 ▶). Accordingly, the two structures superimpose almost exactly, with a Cα r.m.s. deviation of 0.05 Å. Significant differences include the identification of glycerol near one of the copper-binding residues His147, the building of a second conformation of Met170, a residue thought to be involved in Cu2+ reduction, and the presence of a second side-chain conformation of Glu183 in the higher resolution structure.
Figure 1.

A ribbon diagram of CuBD in stereoview. The N- and C-termini are labelled. The α-helix is coloured blue (residues 147–160) and the β-strands are depicted by pink arrows (β1, residues 133–139; β2, 163–174; β3, 177–188). The copper-binding residues His147, His151 and Tyr168 are coloured light blue, blue and gold, respectively. The three disulfides are shown as sticks (from left to right: Cys133–Cys187, Cys158–Cys186 and Cys144–Cys174). The diagram was prepared using PyMOL (DeLano Scientific LLC, USA).
3.3. Water structure
A total of 115 water molecules were identified in the 0.85 Å resolution structure. Of those, 79 water molecules were located in the first hydration shell, which interacts with surface side chains or exposed polar groups of the backbone. A further 24 were located in the second shell and five in the third shell, with some of these water molecules forming an extensive water network linking adjacent symmetry-related CuBD molecules. Another seven appear to be isolated (>4 Å from a hydrogen-bonding partner). There are no internal water molecules in CuBD. Comparing against the lower resolution structure, 95 water sites are found to be in common.
3.4. Analysis of geometric parameters
The atomic resolution model was refined without restraints, except for Glu131, Ala132, His151 (side chain) and Ile176 (side chain) which displayed large deviations (6–9 standard deviations) of bond lengths and angles from ideal values when left unrestrained. This may be an attempt by SHELXL to account for the high mobility of these residues and hence restraints were re-introduced for them.
A list of main-chain bond lengths and bond angles was compiled by the program PROCHECK (Laskowski et al., 1993 ▶) and their values were compared against the Engh and Huber ideal values (Engh & Huber, 1991 ▶). The distributions of bond lengths and angles are summarized in Tables 4 ▶ and 5 ▶, respectively. Overall, the mean and standard deviations of the bond lengths and angles agree well with the ideal values (within 2 standard deviations), apart from the mean N—Cα bond length of glycine residues (2.5 and 3 standard deviations). However, since there are only a small number of glycine residues (three), the latter observation may not be so significant. The mean peptide-bond dihedral angle ω is +179.2° and the standard deviation 6.6°, compared with the normal mean of +178° and standard deviation of 5.5° as defined by the geometry-validation program WHATCHECK (Hooft et al., 1996 ▶).
Table 4. Analysis of bond lengths in the high-resolution CuBD structure compiled by PROCHECK .
Each asterisk represents one standard deviation and each plus sign represents half a standard deviation. Therefore, +*** indicates that the value of the parameter is between 3.5 and 4.0 standard deviations from the Engh and Huber value. Standard deviations greater than 4.5 are listed after the symbol ^.
| Engh & Huber | Sub-angstrom CuBD133–189 structure | ||||||
|---|---|---|---|---|---|---|---|
| Bond | Mean | St. dev. | No. of values | Minimum | Maximum | Mean | St. dev. |
| C—N | |||||||
| Except Pro | 1.329 | 0.014 | 56 | 1.29+** | 1.391**** | 1.333 | 0.018 |
| Pro | 1.341 | 0.016 | 2 | 1.334 | 1.337 | 1.336 | 0.002 |
| C—O | 1.231 | 0.02 | 58 | 1.177+** | 1.297*** | 1.234 | 0.022 |
| Cα—C | |||||||
| Except Gly | 1.525 | 0.021 | 56 | 1.467+** | 1.579+** | 1.519 | 0.021 |
| Gly | 1.516 | 0.018 | 3 | 1.523 | 1.545+* | 1.531 | 0.01 |
| Cα—Cβ | |||||||
| Ala | 1.521 | 0.033 | 2 | 1.468+* | 1.518 | 1.493 | 0.025 |
| Ile, Thr, Val | 1.540 | 0.027 | 9 | 1.485** | 1.574* | 1.543 | 0.025 |
| Others | 1.530 | 0.020 | 45 | 1.436^4.7 | 1.592*** | 1.533 | 0.028 |
| N—Cα | |||||||
| Except Gly, Pro | 1.458 | 0.019 | 54 | 1.396*** | 1.488+* | 1.449 | 0.021 |
| Gly | 1.451 | 0.016 | 3 | 1.363^5.5 | 1.434* | 1.407+** | 0.032 |
| Pro | 1.466 | 0.015 | 2 | 1.466 | 1.484 | 1.475 | 0.009 |
Table 5. Analysis of bond angles in the high-resolution CuBD structure compiled by PROCHECK .
See the legend to Table 4 ▶ for explanation of symbols.
| Engh & Huber | Sub-angstrom CuBD133–189 structure | ||||||
|---|---|---|---|---|---|---|---|
| Angle | Mean | St. dev. | No. of values | Minimum | Maximum | Mean | St. dev. |
| Cα—C—N | |||||||
| Except Gly, Pro | 116.2 | 2.0 | 53 | 112.4+* | 123*** | 116.8 | 2.12 |
| Gly | 116.4 | 2.1 | 3 | 111.58** | 115.74 | 113.39* | 1.74 |
| Pro | 116.9 | 1.5 | 2 | 111.51+*** | 115.54 | 113.52** | 2.02 |
| O—C—N | |||||||
| Except Pro | 123.0 | 1.6 | 56 | 118.41+** | 126.32** | 122.44 | 1.76 |
| Pro | 122.0 | 1.4 | 2 | 123.63* | 124.88** | 124.25+* | 0.62 |
| C—N—Cα | |||||||
| Except Gly, Pro | 121.7 | 1.8 | 53 | 117.29** | 125.88** | 121.91 | 2.2 |
| Gly | 120.6 | 1.7 | 3 | 119.51 | 125.45+** | 122.69* | 2.44 |
| Pro | 122.6 | 5.0 | 2 | 119.55 | 120.08 | 119.82 | 0.27 |
| Cα—C—O | |||||||
| Except Gly | 120.8 | 1.7 | 55 | 117.46+* | 124.52** | 120.84 | 1.52 |
| Gly | 120.8 | 2.1 | 3 | 121.52 | 123.51* | 122.38 | 0.84 |
| Cβ—Cα—C | |||||||
| Ala | 110.5 | 1.5 | 2 | 110.89 | 112.97+* | 111.93 | 1.04 |
| Ile, Thr, Val | 109.1 | 2.2 | 9 | 107.15 | 114.2** | 111.23 | 1.92 |
| The rest | 110.1 | 1.9 | 45 | 104.8+** | 114.66** | 110.43 | 2.12 |
| N—Cα—C | |||||||
| Except Gly, Pro | 111.2 | 2.8 | 54 | 106.38+* | 114.91* | 110.43 | 1.75 |
| Gly | 112.5 | 2.9 | 3 | 108.89* | 111.42 | 109.93 | 1.08 |
| Pro | 111.8 | 2.5 | 2 | 111.75 | 111.94 | 111.84 | 0.1 |
| N—Cα—Cβ | |||||||
| Ala | 110.4 | 1.5 | 2 | 107.75+* | 108.79* | 108.27* | 0.52 |
| Ile, Thr, Val | 111.5 | 1.7 | 9 | 109.23* | 113.21* | 111.21 | 1.37 |
| Pro | 103.0 | 1.1 | 2 | 101.55* | 102.11 | 101.83* | 0.28 |
| Others | 110.5 | 1.7 | 43 | 106.54 | 113.07 | 110.39 | 1.44 |
3.5. Analysis of anisotropy
The displacements of all atoms were determined anisotropically and are illustrated by means of 50% probability vibration ellipsoids in Fig. 2 ▶. The motions of many of the atoms are rather isotropic as their ellipsoids are quite close to spherical. Atoms with lower B factors (increasing blueness in Fig. 2 ▶) tend to exhibit more isotropic motion, whereas those with high B factors (coloured yellow or light green) are generally more anisotropic, as depicted by elongated ellipsoids. The latter are generally found on the periphery of the protein exposed to the surface, particularly the loop joining the α-helix to strand β2 and that joining strands β2 and β3, so it is not surprising that they have greater freedom of motion.
Figure 2.

An illustration of the atomic motions in CuBD. Atomic motions are depicted by means of 50% probability vibration ellipsoids. The ellipsoids are coloured according to isotropic B factors, from blue (B factor of 5 Å2) through green to yellow (B factor exceeding 25 Å2). The figure was prepared with the online program PARVATI (Merritt, 1999 ▶).
3.6. Implications for copper binding and the redox mechanism
The copper-binding residues His147, His151 and Tyr168 in the atomic resolution structure superimpose almost exactly with their counterparts in the previous 1.6 Å resolution apo structure (Figs. 3 ▶ a and 3 ▶ b). There is no evidence of any bound Cu2+. Their maximum anisotropic displacement parameters do not exceed 0.2 Å2, with the mean ranging between 0.12 and 0.17 Å2 in the x, y and z directions. In other words, the atomic motions in those residues do not exceed 0.45 Å (the square root of 0.2). In comparison, the mean displacement parameters in each of the x, y and z directions range from 0.11 to 0.13 Å2 for buried side chains, between 0.12 and 0.15 Å2 for other surface side-chains emanating from defined secondary-structure elements and between 0.17 and 0.21 Å2 for side chains on the surface loops. Hence, the copper-binding residues appear to have limited movement compared with mobile surface residues, even though they are found on the protein surface and are not involved in crystal contacts. This is in accord with the relatively minor movements exhibited by these residues upon Cu binding (Kong et al., 2007 ▶).
Figure 3.

The copper-binding site at atomic resolution (a) and a comparison with the previous 1.6 Å resolution structure (b) and the Cu2+-bound CuBD structure (c). All the figures are presented in stereoview and were prepared using PyMOL (DeLano Scientific LLC, USA). The atomic resolution structure is coloured in the standard atomic colouring scheme. In (a), the blue mesh represents 2F obs − F calc density contoured at 1.5σ. In (b), the 1.6 Å resolution structure is coloured in aqua throughout. In (c), the C atoms of the Cu2+-bound CuBD structure are coloured purple, the Cu2+ ion orange and the water ligands grey.
The water molecule within hydrogen-bonding distance of His151 is more mobile in comparison. The anisotropy value of this water is 0.37 and its maximum anisotropic displacement parameter is 0.53 Å2 (U 11), suggesting atomic motions of about 0.7 Å. Thus, this water molecule does not seem to be strongly bonded to His151. It may have enough flexibility to participate in the coordination of an incoming Cu ion as an ‘equatorial’ ligand (as seen in the previously published structure) or be displaced when the CuBD interacts with other binding partners or other APP domains (Kong et al., 2007 ▶). The apical water involved in Cu2+ coordination is not observed in the atomic resolution structure.
The normal biological role of copper binding by APP is still not clear. For example, CuBD could be involved in copper transport (White, Reyes et al., 1999 ▶), modulation of APP processing (Borchardt et al., 2000 ▶) or catalysing reactions that are driven by switches in the Cu redox state, such as the autoprocessing of the heparan sulfate chains in glypican-1 (Cappai et al., 2005 ▶). The identification of a glycerol molecule near the copper-binding residue His147 may be suggestive of where and how a potential ligand can bind to CuBD.
Although Met170 is not involved directly in binding Cu ions, it may still play a role in driving Cu2+ reduction (White, Multhaup et al., 1999 ▶). It is of interest that alternative conformations of Met170 are observed in the present structure. The occupancies of the two conformations are 0.65 and 0.35. In the former the thioether is about 7 Å from the copper-binding site and points away from the other copper-binding residues, while in the second conformation the thioether group is closer to the copper-binding site (6 Å) and points towards the Cu ion, although Met170 itself is still buried away from the surface (Fig. 3 ▶ c). The flexibility of the Met170 side chain may be important in facilitating Cu-ion binding or the reduction of Cu2+ to Cu+ after Cu2+ has bound to the CuBD (White et al., 2002 ▶; Barnham et al., 2003 ▶).
The ultrahigh-resolution structure of CuBD presented here provides an excellent opportunity for structure-based drug design of small molecules that might mimic the effects of copper binding to APP, which causes depletion of Aβ production.
Supplementary Material
PDB reference: Alzheimer’s disease amyloid precursor protein copper-binding domain, 2fma, r2fmasf
Acknowledgments
We wish to thank Denise Galatis and William McKinstry for expressing the CuBD and for advice on protein purification and crystallization, respectively. We thank Professor Hans Freeman for advice and encouragement. Technical assistance in data collection was provided by Michael Bolbat and other staff at BioCARS (Beamline 14) of the Advanced Photon Source synchrotron. This work, including the use of the BioCARS sector, was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Energy Research. This work was also supported by grants from the National Health and Medical Research Council of Australia (NHMRC) to MWP and RC. RC is an NHMRC Senior Research Fellow and MWP is an Australian Research Council Federation Fellow and NHMRC Honorary Fellow. GK-WK was an Australian Postgraduate Award Scholar and a recipient of a Ludo Frevel Scholarship from the International Centre for Diffraction Data.
References
- Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., Williamson, N. A., White, A. R., Hinds, M. G., Norton, R. S., Beyreuther, K., Masters, C. L., Parker, M. W. & Cappai, R. (2003). J. Biol. Chem.278, 17401–17407. [DOI] [PubMed]
- Bayer, T. A., Schäfer, S., Simons, A., Kemmling, A., Kamer, T., Tepest, R., Eckert, A., Schüssel, K., Eikenberg, O., Sturchler-Pierrat, C., Abramowski, D., Staufenbiel, M. & Multhaup, G. (2003). Proc. Natl Acad. Sci. USA, 100, 14187–14192. [DOI] [PMC free article] [PubMed]
- Borchardt, T., Camakaris, J., Cappai, R., Masters, C. L., Beyreuther, K. & Multhaup, G. (1999). Biochem. J.344, 461–467. [PMC free article] [PubMed]
- Borchardt, T., Schmidt, C., Camakaris, J., Cappai, R., Masters, C. L., Beyreuther, K. & Multhaup, G. (2000). Cell. Mol. Biol.46, 785–795. [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Cappai, R., Cheng, F., Ciccotosto, G. D., Needham, B. E., Masters, C. L., Multhaup, G., Fransson, L.-Å. & Mani, K. (2005). J. Biol. Chem.280, 13919–13920. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Esch, F. S., Keim, P. S., Beattie, E. C., Blacher, R. W., Culwell, A. R., Oltersdorf, T., McClure, D. & Ward, P. J. (1990). Science, 248, 1122–1125. [DOI] [PubMed]
- Haass, C. & Selkoe, D. J. (2007). Nature Rev. Mol. Cell Biol.8, 101–112. [DOI] [PubMed]
- Harrison, P. M. & Sternberg, M. J. E. (1994). J. Mol. Biol.244, 448–463. [DOI] [PubMed]
- Hooft, R. W. W., Vriend, G., Sander, C. & Abola, E. E. (1996). Nature (London), 381, 272. [DOI] [PubMed]
- Iwatsubo, T. (2004). Curr. Opin. Neurobiol.14, 378–383.
- Kang, J., Lemaire, H.-G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K.-H., Multhaup, G., Beyreuther, K. & Müller, U. (1987). Nature (London), 325, 733–736. [DOI] [PubMed]
- Kleywegt, G. J. & Jones, T. A. (2002). Structure, 10, 465–472. [DOI] [PubMed]
- Klyubin, I., Walsh, D. M., Lemere, C. A., Cullen, W. K., Shankar, G. M., Betts, V., Spooner, E. T., Jiang, L., Anwyl, R., Selkoe, D. J. & Rowan, M. J. (2005). Nature Med.11, 556–561. [DOI] [PubMed]
- Kong, G. K.-W., Adams, J. J., Harris, H. H., Boas, J. F., Curtain, C. C., Galatis, D., Masters, C. L., Barnham, K. J., McKinstry, W. J., Cappai, R. & Parker, M. W. (2007). J. Mol. Biol.367, 148–161. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., Gallagher, M. & Ashe, K. H. (2006). Nature (London), 440, 352–357. [DOI] [PubMed]
- Maynard, C. J., Bush, A. I., Masters, C. L., Cappai, R. & Li, Q.-X. (2005). Int. J. Exp. Pathol.86, 147–159. [DOI] [PMC free article] [PubMed]
- Merritt, E. A. (1999). Acta Cryst. D55, 1109–1117. [DOI] [PubMed]
- Multhaup, G., Schlicksupp, A., Hesse, L., Beher, D., Ruppert, T., Masters, C. L. & Beyreuther, K. (1996). Science, 271, 1406–1409. [DOI] [PubMed]
- Nishimoto, I., Okamoto, T., Matsuura, Y., Okamoto, T., Murayama, Y. & Ogata, E. (1993). Nature (London), 362, 75–79. [DOI] [PubMed]
- Okamoto, T., Takeda, S., Murayama, Y., Ogata, E. & Nishimoto, I. (1995). J. Biol. Chem.270, 4205–4208. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Phinney, A. L. et al. (2003). Proc. Natl Acad. Sci. USA, 100, 14193–14198.
- Richardson, J. S. (1981). Adv. Protein Chem.34, 167–339. [DOI] [PubMed]
- Rossjohn, J., Cappai, R., Feil, S. C., Henry, A., McKinstry, W. J., Galatis, D., Hesse, L., Multhaup, G., Beyreuther, K., Masters, C. L. & Parker, M. W. (1999). Nature Struct. Biol.6, 327–331. [DOI] [PubMed]
- Schmidt, A. & Lamzin, V. S. (2002). Curr. Opin. Struct. Biol.212, 698–703. [DOI] [PubMed]
- Schneider, M. (1996). Proceedings of the CCP4 Study Weekend. Macromolecular Refinement, edited by E. Dodson, M. Moore, A. Ralph & S. Bailey, pp. 133–144. Warrington: Daresbury Laboratory.
- Schüttelkopf, A. W. & van Aalten, D. M. F. (2004). Acta Cryst. D60, 1355–1363. [DOI] [PubMed]
- Selkoe, D. J. (2002). Science, 298, 789–791. [DOI] [PubMed]
- Sheldrick, G. M. (1997). SHELX97. Programs for Crystal Structure Analysis (Release 97-2). University of Göttingen, Germany.
- Sinha, S. et al. (1999). Nature (London), 402, 537–540.
- Vrielink, A. & Sampson, N. (2003). Curr. Opin. Struct. Biol.13, 709–715. [DOI] [PubMed]
- White, A. R., Multhaup, G., Galatis, D., McKinstry, W. J., Parker, M. W., Pipkorn, R., Beyreuther, K., Masters, C. L. & Cappai, R. (2002). J. Neurosci.22, 365–376. [DOI] [PMC free article] [PubMed]
- White, A. R., Multhaup, G., Maher, F., Bellingham, S., Camakaris, J., Zheng, H., Bush, A. I., Beyreuther, K., Masters, C. L. & Cappai, R. (1999). J. Neurosci.19, 9170–9179. [DOI] [PMC free article] [PubMed]
- White, A. R., Reyes, A. E., Mercer, J. F. B., Camakaris, J., Zheng, H., Bush, A. I., Multhaup, G., Beyreuther, K., Masters, C. L. & Cappai, R. (1999). Brain Res.842, 439–444. [DOI] [PubMed]
- Yan, R., Blenkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., Brashler, J. R., Stratman, N. C., Mathews, W. R., Buhl, A. E., Carter, D. B., Tomasselli, A. G., Parodi, L. A., Heinrikson, R. L. & Gurney, M. E. (1999). Nature (London), 402, 533–536. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Alzheimer’s disease amyloid precursor protein copper-binding domain, 2fma, r2fmasf