

Binding of Ni2+ ions to the uncleaved affinity tag facilitated de novo phasing of the crystal structure of M. tuberculosis mtFabD to 3.0 Å resolution.
Keywords: mtFabD, malonyl-CoA:acyl carrier protein transacylases
Abstract
Mycobacteria display a unique and unusual cell-wall architecture, central to which is the membrane-proximal mycolyl-arabinogalactan-peptidoglycan core (mAGP). The biosynthesis of mycolic acids, which form the outermost layer of the mAGP core, involves malonyl-CoA:acyl carrier protein transacylase (MCAT). This essential enzyme catalyses the transfer of malonyl from coenzyme A to acyl carrier protein AcpM, thus feeding these two-carbon units into the chain-elongation cycle of the type II fatty-acid synthase. The crystal structure of M. tuberculosis mtFabD, the mycobacterial MCAT, has been determined to 3.0 Å resolution by multi-wavelength anomalous dispersion. Phasing was facilitated by Ni2+ ions bound to the 20-residue N-terminal affinity tag, which packed between the two independent copies of mtFabD.
1. Introduction
Mycobacterium tuberculosis remains the single most important bacterial cause of mortality and morbidity in the world, with around 8 million cases and 3 million deaths annually (Dye et al., 2005 ▶). Factors such as HIV/AIDS, multi-drug resistance and the variable efficacy of the BCG vaccine have compounded a problem that, since the discovery of the tubercle bacillus by Koch in the late nineteenth century, has remained constant in its global dimensions. There is a clear need to extend our understanding of the physiology and pathogenicity of Mycobacterium tuberculosis in the hope of priming novel immunoprophylactic and therapeutic approaches. Mycobacteria and related taxa are unique in that they feature a highly impermeable hydrophobic cell wall consisting of solvent-extractable lipids, glycans and proteins that are interspersed into the membrane-proximal, covalently linked mycolyl-arabinogalactan-peptidoglycan core (mAGP). The outermost layer of the mAGP core is made of mycolic acids, very long α-alkyl β-hydroxylated fatty acids that appear also as trehalose conjugates in the outer segment and are essential for viability (Takayama et al., 2005 ▶).
Mycolic acids are composed of a characteristic meromycolate chain (C54–60) and a shorter C24–26 α-alkyl chain. The synthesis of the meromycolate chain involves both type I and type II fatty-acid synthases (FAS). FAS-I generates C16–24 alkyl chains from acetyl-CoA and malonyl-CoA precursors by way of cyclic two-carbon elongation. The resulting medium-chain fatty acids are further extended by the FAS-II system, a reversible complex of enzymes encoded by separate genes, which catalyses further (two-carbon cyclic) elongation, yielding C54–60 chains (Takayama et al., 2005 ▶). After release from FAS-II, the meromycolate and α-alkyl chains are fused by way of a Claisen-type condensation reaction. This essential step is catalysed by a type-1 polyketide synthase, Pks13 (Rv3800c; Portevin et al., 2004 ▶).
FAS-II acts solely on acyl-chain substrates bound to the phosphopantetheine arm of acyl-carrier protein AcpM; hence, coenzyme A (CoA) linked fatty acids first need to undergo transacylation from CoA to AcpM. Transacylation of the medium-chain CoA derivatives released by FAS-I is catalysed by the β-ketoacyl ACP synthase III FabH (Brown et al., 2005 ▶; Choi et al., 2000 ▶), while the malonyl-CoA:ACP transacylase (MCAT) mtFabD facilitates entry of the malonyl extender units into the FAS-II system and constitutes a serine-dependent acylhydrolase catalysing transacylation by a ping-pong Bi-Bi kinetic reaction mechanism (Joshi & Wakil, 1971 ▶). FabD has been shown to be essential (Zhang & Cronan, 1998 ▶) and can be regarded as an attractive drug target (Zhang et al., 2006 ▶). To date, only one inhibitor of MCAT activity has been identified (Liu et al., 2006 ▶). In order provide a more solid basis for rational drug design targeting M. tuberculosis mtFabD, we determined its crystal structure in a two-wavelength anomalous dispersion experiment.
2. Experimental
2.1. Protein purification and crystallization
All reagents were sourced from Sigma–Aldrich, unless stated otherwise. Escherichia coli C41(DE3) cells (Avidis) were transformed with plasmid pET28(a)-mtfabD as described in Kremer et al. (2001 ▶) and grown on agar plates containing 25 µg ml−1 kanamycin. We note that the pET28 vector includes a 20-residue N-terminal tag containing a His6 sequence, −19MGSSHHHHHHSSGLVPRGSH0 (single-letter code with superscripts denoting residue numbers relative to the initial methionine of the FabD sequence), which was not enzymatically removed during the preparation. An overnight culture of LB medium containing 25 µg ml−1 kanamycin (310 K, 180 rev min−1) was inoculated from a single colony and used to inoculate 1 l Terrific Broth (Sambrook et al., 1998 ▶). The bulk culture was grown at 310 K until the OD600 reached 0.6 and then allowed to cool; it was then induced with 1 mM isopropyl β-d-thiogalactoside (IPTG), followed by incubation at 289 K (16 h). Cells were harvested and the pellet was resuspended in column buffer (20 mM sodium phosphate pH 7.9, 500 mM NaCl) and cells were lysed using a French press. Lysates were centrifuged (27 000g, 277 K) and the supernatant was loaded onto an Ni–NTA column (Amersham HiTrap, 5 ml) equilibrated in column buffer. MtFabD was eluted using column buffer with imidazole (25–500 mM) and the fractions were analysed by SDS–PAGE. Pure fractions were pooled and dialysed into storage buffer (50 mM NaCl, 20 mM Tris–HCl pH 7.9) and concentrated to ∼20 mg ml−1 using Centriprep/Centricon ultrafiltration columns (Millipore). At this point malonyl-CoA was added at a molar ratio of 4:1 (malonyl-CoA:protein) and ultrafiltration was resumed to a final protein concentration of ∼50 mg ml−1.
Crystals of mtFabD were obtained by hanging-drop vapour diffusion, initially against sparse-matrix screens (Molecular Dimensions Ltd), mixing 1 µl protein with 1 µl reservoir solution. Initial crystals appeared in 20% PEG MME 2000, 100 mM Tris–HCl pH 8.5 and 10 mM NiCl2. Optimized crystals (150 × 150 × 50 µm) suitable for X-ray data collection were grown over a reservoir containing 10–12% PEG MME 2000, 100 mM Tris–HCl pH 8.0–8.5 and 5–10 mM NiCl2.
2.2. X-ray data acquisition, structure determination and refinement
Prior to data collection, crystals were soaked in reservoir solution containing increasing concentrations of glycerol (5% steps, 90 s per step, followed by 5 min in the final step) to a maximum of 30%, mounted in nylon loops, frozen and stored in liquid nitrogen. X-ray data were recorded on beamline ID23-1 at ESRF, Grenoble, France. Crystals belonged to space group P6222 with two molecules per crystallographic asymmetric unit. Crystals were observed to suffer significant radiation damage at the Ni K-edge peak; hence, data were first recorded at a remote wavelength (12.5 keV), followed by data acquisition at the absorption peak (8.346 keV). The data were integrated and reduced using XDS and XSCALE (Kabsch, 1993 ▶; Table 1 ▶) and five Ni2+ ions were located using SHELXD (Schneider & Sheldrick, 2002 ▶) as implemented in autoSHARP (Vonrhein et al., 2006 ▶) using the ‘peak’ and ‘remote’ wavelength data. Heavy-atom positions were refined and phases were calculated using SHARP (de La Fortelle & Bricogne, 1997 ▶; Table 1 ▶), followed by solvent flattening using SOLOMON (Abrahams & Leslie, 1996 ▶). The resulting electron-density map (Fig. 1 ▶ b) was of a very good quality, allowing us to manually place secondary-structure fragments using Coot (Emsley & Cowtan, 2004 ▶), which then served as a guide to unequivocally place two copies of Streptomyces coelicolor A3(2) FabD (PDB code 1nm2; Keatinge-Clay et al., 2003 ▶) into density. Several rounds of rebuilding of this initial model were interspersed with conjugate-gradient minimization in REFMAC5 (Murshudov et al., 1997 ▶) applying phase restraints, but omitting noncrystallographic symmetry (NCS) restraints. The final model comprises two copies of mtFabD covering the complete sequence (residues 1–302), 18 of the 20 residues of the purification tag, five Ni2+ ions and 43 water molecules. Refinement statistics are presented in Table 1 ▶.
Table 1. Data-collection, phasing and structure-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Remote | Peak |
|---|---|---|
| Wavelength (Å) | 0.992 | 1.486 |
| Space group | P6222 | P6222 |
| Unit-cell parameters (Å) | a = 180.2, c = 96.0 | a = 180.2, c = 96.0 |
| Resolution range (Å) | 48–3.0 (3.16–3.0) | 48–3.0 (3.16–3.0) |
| Unique reflections | 18818 (2606) | 18886 (2686) |
| Completeness (%) | 99.9 (99.5) | 99.9 (100.0) |
| Rsym† (%) | 9.0 (34.0) | 10.5 (58.1) |
| Multiplicity | 13.0 (13.2) | 7.3 (7.4) |
| 〈I〉/〈σ(I)〉 | 22.2 (7.8) | 13.3 (3.2) |
| Figure of merit‡ (acentric/centric) | 0.746/0.819 | |
| Phasing power (30–3.0 Å) | 0.579 | |
| No. of protein atoms | 4548 | |
| No. of Ni2+ ions | 5 | |
| No. of water molecules | 43 | |
| Rcryst§ | 22.3 | |
| Rfree§ | 26.7 | |
| R.m.s.d. bonds (Å) | 0.008 | |
| R.m.s.d. angles (°) | 1.2 | |
| R.m.s.d. B of bonded atoms (Å2) | ||
| Main chain | 0.30 | |
| Side chain | 0.81 | |
| Average B factor (Å2) | 64 | |
| Wilson B factor (Å2) | 72 | |
R
sym = 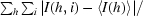
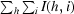 , where I(h, i) is the intensity of the ith measurement of reflection h and 〈I(h)〉 is the mean value of I(h, i) for all i measurements.
, where I(h, i) is the intensity of the ith measurement of reflection h and 〈I(h)〉 is the mean value of I(h, i) for all i measurements.
Figure of merit after phase calculation in SHARP and before solvent flattening.
R
cryst = 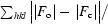
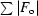 , where F
o is the observed structure-factor amplitude and F
c the calculated structure-factor amplitude. R
free is calculated based on 5% of reflections not used in refinement.
, where F
o is the observed structure-factor amplitude and F
c the calculated structure-factor amplitude. R
free is calculated based on 5% of reflections not used in refinement.
Figure 1.

Ribbon diagram of M. tuberculosis mtFabD and experimental density. (a) The large and small subdomains are highlighted in green and magenta, respectively. The N-terminal affinity tag is shown as a stick model, with Ni2+ ions appearing as blue spheres. The location of the active site is indicated by a stick model of CoA, based on the superposition with the structure of E. coli FabD in complex with CoA and malonate (PDB code 2g2z; Oefner et al., 2006 ▶). (b) Close-up view of the affinity tag in the superposition of the two independent copies of mtFabD with respect to the Cα atoms of residues 1–302, i.e. excluding the affinity tag. Molecules A and B are shown in pale red and cyan, respectively. Experimental density corresponding to molecule B contoured at 1.2σ is shown, but is restricted to the vicinity of the affinity tag for clarity. All figures were generated using PyMOL v.0.97 (DeLano Scientific LLC).
3. Results and discussion
3.1. Structure of mtFabD
The structure of M. tuberculosis mtFabD was determined by two-wavelength anomalous dispersion to 3.0 Å resolution (Table 1 ▶) and represents the (affinity-tagged) enzyme previously characterized by us (Kremer et al., 2001 ▶). Experimental phases were obtained based on the anomalous scattering of the ordered Ni2+ ions bound by the histidine residues of the uncleaved purification tag (Fig. 1 ▶). Omission of Ni2+ from the crystallization conditions failed to produce crystals. Similarly, the production of well diffracting crystals was dependent on the presence of the substrate malonyl-CoA during the final stages of ultrafiltration. However, no density attributable to malonyl-CoA was visible. Despite the limited resolution the experimental density map was of a very good quality (Fig. 1 ▶ b), allowing us to build the entire structure into experimental density, relying on calculated density maps only at later stages of the refinement.
The fold of M. tuberculosis mtFabD is identical to that of other MCAT enzymes. Structures of bacterial orthologues are available from E. coli (Oefner et al., 2006 ▶; Serre et al., 1995 ▶), S. coelicolor (Keatinge-Clay et al., 2003 ▶) and more recently from Helicobacter pylori (Zhang et al., 2007 ▶), in addition to a structure of the MCAT domain of human fatty-acid synthase (PDB code 2jfk; G. Bunkoczi, K. Kavanagh, V. Hozjan, A. Rojkova, S. Watt, X. Wu, C. H. Arrowsmith, A. Edwards, M. Sundstrom, J. Weigelt, S. Smith & U. Oppermann, unpublished work). At the time of completing this report, a structure of M. tuberculosis mtFabD had been deposited in the PDB but not yet released (Li et al., 2007 ▶). Superposition with these structures highlights the high level of evolutionary conservation of this essential enzyme, as is evident by root-mean-square deviations (r.m.s.d.s) below 2.0 Å: S. coelicolor (PDB code 1nm2), r.m.s.d. 1.02 Å for 290 aligned Cα atoms; E. coli (Fig. 2 ▶ a; PDB code 2g2z), r.m.s.d. 1.34 Å for 255 Cα pairs; MCAT domain of human fatty-acid synthase (Fig. 2 ▶ b; PDB code 2jfk), r.m.s.d. 1.46 Å for 217 Cα pairs. Interestingly, a DALI search (http://www.ebi.ac.uk/dali; Holm & Sander, 1999 ▶) revealed a clear gap in structural similarity between transacylases and more distantly related structures: the two top hits (E. coli FabD and the MCAT domain of human FAS) scored Z values above 30, whereas the third entry (nonspecific lipid acyl hydrolase patatin; Rydel et al., 2003 ▶) scored 6.5 on a scale that considers hits with scores of 2 or less to be unrelated structures. Nevertheless, similarities in fold between MCAT enzymes and α/β-hydrolases have been noted previously (Keatinge-Clay et al., 2003 ▶).
Figure 2.

Comparison of M. tuberculosis MtFabD (molecule B in cyan) with orthologous MCAT enzymes (in grey). Superposition of M. tuberculosis MtFabD (a) with E. coli FabD (PDB code 2g2z; Oefner et al., 2006 ▶) and (b) with the MCAT domain of human fatty-acid synthase (PDB code 2jfk).
MtFabD comprises two subdomains (Fig. 1 ▶ a). The large subdomain is split into two noncontiguous segments (residues 1–126 and 199–302) and consists of a parallel five-stranded β-sheet sandwiched between a seven-helix bundle and two shorter helices (Fig. 1 ▶ a). The small subdomain, spanning residues 127–198, forms a lid-like cover over the active site and consists a four-stranded antiparallel β-sheet with two helices stacked on top. Both subdomains contribute to the active site and carry residues critical for the catalytic reaction mechanism.
The active site is located in the cleft between the small and large subdomains. Key catalytic elements include the side chains of Ser91, Arg116 and His194 and the backbone amide of Gln9 (Fig. 3 ▶). Ser91 and His194 form a catalytic dyad, with His194 acting as a base abstracting the proton from the hydroxyl of the catalytic serine (Keatinge-Clay et al., 2003 ▶). The latter is located in the so-called catalytic elbow, a structural motif typically found in α/β-hydrolases. The acyl carbonyl is thought to bind into an oxyanion hole, to which the amide N atom of Gln9 contributes (Fig. 3 ▶). The guanido group of Arg116 helps to position the malonyl-CoA with respect to the catalytic dyad by forming a salt bridge with the malonyl carboxylate (Keatinge-Clay et al., 2003 ▶). In a previous study, we showed that mutations of Ser91, His194, Arg116 result in drastic reductions in (if not complete abolition of) transacylase activity of M. tuberculosis mtFabD (Kremer et al., 2001 ▶). Although neither the side chain of Gln9 nor that of Gln243 is thought to be involved in contacting malonyl-CoA, their mutation has a similarly deleterious effect on enzymatic activity. The imidazole ring of the catalytic histidine is observed to be within hydrogen-bonding distance of the carbonyl of Gln243 (Fig. 3 ▶). Mutations at this site might result in adjustments of the backbone geometry in the region of residue 243, affecting the hydrogen bond to His194, and may thus explain the effect of mutating this site. Analogous considerations are likely to apply to Gln9.
Figure 3.

Close-up view of the active site of M. tuberculosis mtFabD. Amino acids critical for catalysis are shown as sticks and coloured by element (yellow, carbon; red, oxygen; blue, nitrogen). The stick model in grey indicates the putative position of coenzyme A derived from the structure of E. coli FabD in complex with malonate and CoA (PDB code 2g2z; Oefner et al., 2006 ▶). Selected hydrogen bonds are drawn as dashed lines.
3.2. Ordered affinity tag
The asymmetric unit comprises two independent molecules of mtFabD, which were built independently and not restrained by NCS. The two molecules are related by a pseudo-twofold rotation (Fig. 4 ▶) with direction cosines (0.31, −0.95, −0.1). The two polypeptide chains superimpose with an r.m.s.d. of 0.48 Å for 302 paired Cα atoms. The uncleaved 20-residue affinity tag resulting from the expression vector (see §2.1) was ordered: in molecule B the entire tag was visible, only lacking the two N-terminal residues, while in molecule A a gap spanning residues −7 to −2 was observed in the tag, while the Ni2+-bound His residues −10 through −17 were well defined in the density (Fig. 1 ▶ b). The ordering of the purification tag was a result of it forming a packing interface between the two independent molecules of the asymmetric unit. The five heavy-atom sites are symmetrically arranged around the NCS twofold rotation axis, with site 4 located on the axis and sites 1 and 3 being NCS-related to sites 5 and 2, respectively (Fig. 4 ▶ a). Nickel site 4 is coordinated by histidine residues −15 and −13 of both chains (Fig. 4 ▶ a). Two main-chain hydrogen bonds (3.0 and 3.1 Å in length; Fig. 4 ▶ a) between the His residues at position −15 provide additional stabilizing contacts between the two chains. In addition to the His residues of the tag, the Ni2+ ions are coordinated by Ser−17 (sites 1, 5) and by Asp84 and Asp216 (the only nontag residues to do so), coordinating sites 2 and 3, which are closest to the initial Met residue of the mtFabD sequence. Residues 210–218 and Ala233 make contacts with tag residues of the opposite monomer (Fig. 4 ▶ a), contributing to an apparent dimer interface that is situated on the ‘back’ of the enzyme, about 27 Å away from the active site (Fig. 4 ▶ b). Importantly, there are no direct contacts between residues of the actual mtFabD sequence across this interface and we consider the tag-mediated dimerization to be a crystallization artefact. Indeed, in sedimentation-velocity experiments the His-tagged mtFabD migrated as a monomer (A. K. Brown & G. S. Besra, unpublished data). The affinity tag orders on the ‘back’ of the enzyme, not interfering with the active site (Fig. 4 ▶ b) and forming a 180° turn in its conformation (Fig. 1 ▶). The 180° turn is the result of His0, the residue immediately upstream of the initial methionine, participating in the coordination of nickel sites 3 and 2 in chains A and B, respectively (Fig. 4 ▶ a). The identical conformation of the affinity tags in molecules A and B (Fig. 1 ▶ b), notwithstanding the disordered region in chain A, seems remarkable, but mirrors the twofold symmetric arrangement of the five nickel ions (Fig. 4 ▶ a).
Figure 4.

Affinity-tag-mediated interface between molecules A (pale red) and B (cyan) of the asymmetric unit, with tag residues in stick representation and Ni2+ ions as blue spheres. (a) Close-up view, with the ribbon in yellow and magenta representing residues 210–218, which make contacts with the tag of the opposite monomer. Yellow dashed lines indicate the main-chain hydrogen bonds between the NCS-related copies of His−15. The NCS twofold axis is approximately parallel to the viewing direction and runs through nickel site 4 (labels in magenta). (b) View perpendicular to (a), demonstrating that the (pseudo)-dimer interface is located distal to the active site (indicated for molecule B by the sphere model of CoA). Selected residues are labelled with subscripts denoting the polypeptide chain.
Supplementary Material
PDB reference: mtFabD, 2qj3, r2qj3sf
Acknowledgments
We thank ESRF Grenoble for access to the synchrotron and travel support and the ESRF staff, in particular Andrew McCarthy, for support during data acquisition. GSB acknowledges support in the form of a Personal Research Chair from Mr James Bardrick, as a former Lister Institute–Jenner Research Fellow, and the Medical Research Council. LK is supported by a Grant from the Centre National de la Recherche Scientifique (CNRS; Action Thématique Iniciative sur Programme ‘Microbiologie Fondamentale’).
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Brown, A. K., Sridharan, S., Kremer, L., Lindenberg, S., Dover, L. G., Sacchettini, J. C. & Besra, G. S. (2005). J. Biol. Chem.280, 32539–32547. [DOI] [PubMed]
- Choi, K. H., Kremer, L., Besra, G. S. & Rock, C. O. (2000). J. Biol. Chem.275, 28201–28207. [DOI] [PubMed]
- Dye, C., Watt, C. J., Bleed, D. M., Hosseini, S. M. & Raviglione, M. C. (2005). JAMA, 293, 2767–2775. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Holm, L. & Sander, C. (1999). Nucleic Acids Res.27, 244–247. [DOI] [PMC free article] [PubMed]
- Joshi, V. C. & Wakil, S. J. (1971). Arch. Biochem. Biophys.143, 493–505. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Keatinge-Clay, A. T., Shelat, A. A., Savage, D. F., Tsai, S. C., Miercke, L. J., O’Connell, J. D. III, Khosla, C. & Stroud, R. M. (2003). Structure, 11, 147–154. [DOI] [PubMed]
- Kremer, L., Nampoothiri, K. M., Lesjean, S., Dover, L. G., Graham, S., Betts, J., Brennan, P. J., Minnikin, D. E., Locht, C. & Besra, G. S. (2001). J. Biol. Chem.276, 27967–27974. [DOI] [PubMed]
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Li, Z., Huang, Y., Ge, J., Fan, H., Zhou, X., Li, S., Bartlam, M., Wang, H., Rao, Z. (2007). J. Mol. Biol.371, 1075–1083. [DOI] [PubMed]
- Liu, W., Han, C., Hu, L., Chen, K., Shen, X. & Jiang, H. (2006). FEBS Lett.580, 697–702. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Oefner, C., Schulz, H., D’Arcy, A. & Dale, G. E. (2006). Acta Cryst. D62, 613–618. [DOI] [PubMed]
- Portevin, D., De Sousa-D’Auria, C., Houssin, C., Grimaldi, C., Chami, M., Daffé, M. & Guilhot, C. (2004). Proc. Natl Acad. Sci. USA, 101, 314–319. [DOI] [PMC free article] [PubMed]
- Rydel, T. J., Williams, J. M., Krieger, E., Moshiri, F., Stallings, W. C., Brown, S. M., Pershing, J. C., Purcell, J. P. & Alibhai, M. F. (2003). Biochemistry, 42, 6696–6708. [DOI] [PubMed]
- Sambrook, J., Fritsch, E. F. & Maniatis, T. (1998). Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press.
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed]
- Serre, L., Verbree, E. C., Dauter, Z., Stuitje, A. R. & Derewenda, Z. S. (1995). J. Biol. Chem.270, 12961–12964. [DOI] [PubMed]
- Takayama, K., Wang, C. & Besra, G. S. (2005). Clin. Microbiol. Rev.18, 81–101. [DOI] [PMC free article] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Zhang, L., Liu, W., Xiao, J., Hu, T., Chen, J., Chen, K., Jiang, H. & Shen, X. (2007). Protein Sci.16, 1184–1192. [DOI] [PMC free article] [PubMed]
- Zhang, Y. & Cronan, J. E. Jr (1998). J. Bacteriol.180, 3295–3303. [DOI] [PMC free article] [PubMed]
- Zhang, Y. M., White, S. W. & Rock, C. O. (2006). J. Biol. Chem.281, 17541–17544. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: mtFabD, 2qj3, r2qj3sf