

The cloning, purification and crystallization of YnaF from S. typhimurium are reported along with preliminary X-ray crystallographic studies.
Keywords: universal stress proteins, adenosine 5′-triphosphate (ATP), Salmonella typhimurium
Abstract
The universal stress protein UspF (YnaF) is a small cytoplasmic bacterial protein. The expression of stress proteins is enhanced when cells are exposed to heat shock, nutrition starvation and certain other stress-inducing agents. YnaF promotes cell survival during prolonged exposure to stress and may activate a general mechanism for stress endurance. This manuscript reports preliminary crystallographic studies on YnaF from Salmonella typhimurium. The gene coding for YnaF was cloned and overexpressed and the protein was purified by Ni–NTA affinity chromatography. Purified YnaF was crystallized using vapour-diffusion and microbatch methods. The crystals belong to space group P21, with unit-cell parameters a = 37.51, b = 77.18, c = 56.34 Å, β = 101.8°. A data set was collected to 2.5 Å resolution with 94.6% completeness using an image-plate detector system mounted on a rotating-anode X-ray generator. Attempts to determine the structure are in progress.
1. Introduction
Several stress-related proteins are found in bacteria, archaea, fungi and eukarya. Stress induces or alters the activities of various classes of proteins, including transcription factors, enzymes, molecular chaperones, ion channels and transporters. Induction of these proteins is controlled by a battery of genes and their intricate regulation helps the system to overcome the unfavourable conditions of stress. However, the precise mechanism of cellular protection has not been clearly elucidated. Universal stress proteins (Usps) are induced by stress caused by starvation of carbon, nitrogen, phosphorus, sulfur or amino acids and by exposure to heat, oxidants, metals, H2O2, CdCl2, antibiotics or DNA-damaging agents (Nystrom & Neidhardt, 1992 ▶, 1994 ▶). Usps reprogram the cell towards defence against stress. The expression of some genes responds to the arrest of bacterial growth in general and the corresponding gene products become the most abundant proteins in the stationary phase of the bacterial cell (Nystrom & Neidhardt, 1993 ▶; Gustavsson et al., 2002 ▶). The functions provided by these proteins are essential in bacterial pathogenesis (Liu et al., 2007 ▶). The importance of Usp genes in the bacterial life cycle and in virulence can be studied by creating gene-knockout mutants.
Most organisms are equipped with several Usp genes that either encode small Usp proteins of 14–15 kDa consisting of a single domain (UspF, UspG and UspA) or larger proteins in which two Usp domains occur in tandem (UspE). A few Usp domains occur with other domains, as in the sensor kinase KdpD (Walderhaug et al., 1992 ▶). The structure of a member of the UspA family from Methanocaldococcus jannaschii has been solved by crystallography and revealed the protein to consist of ATP-binding homodimers (Zarembinski et al., 1998 ▶). Usp structures have also been determined from other sources: Haemophilus influenza (PDB code 1jmv), Aquifex aeolias (1q77), Nitrosomonas europaea (2pfs), Thermus thermophilus (1wjg) and the eukaryotic Arabidopsis thaliana (2gm3). H. influenza UspA was found to share 68% sequence identity with E. coli UspA, 12% identity with MJ0577 (M. jannaschii UspA; Sousa & McKay, 2001 ▶) and 20% with Salmonella typhimurium UspF. The H. influenza UspA protein shows no evidence of ATP binding (Sousa & McKay, 2001 ▶), whereas M. jannaschii UspA contains an ATP-binding motif (Zarembinski et al., 1998 ▶). Thus, the Usp protein family can be segregated into at least two subfamilies: one that binds ATP and has an ATP-dependent function and one that does not bind ATP (Sousa & McKay, 2001 ▶). No structures of any Usps belonging to the UspF subfamily have been determined to date. Here, we report the cloning, purification and crystallization of YnaF from S. typhimurium and preliminary X-ray crystallographic studies.
2. Materials and methods
2.1. Cloning
The gene coding for YnaF was PCR-amplified from S. enterica serovar typhimurium genomic DNA using Deep Vent DNA polymerase (New England Biolabs). After amplification of the target gene by sense (GCTCATATGGCTAGCATGAACAGAACGATTC) and antisense (CGGGATCCTTACTCGAGTTAGCGTACCACCAG) primers corresponding to the 5′ and 3′ sequences of the ynaF gene, the PCR-amplified fragment was digested with NheI and BamHI. The fragment corresponds to the gene encoding residues 1–144 of YnaF. It was then cloned into the vector pRSET-C (Invitrogen) encoding an N-terminal hexahistidine tag to facilitate purification. Hence, the expressed protein contains 14 additional amino acids from the vector, including six histidines at the N-terminus, resulting in a protein of molecular weight 17.3 kDa.
The isolation of genomic DNA and plasmid DNA for cloning were carried out according to the method of Sambrook & Russell (2001 ▶). The sequence of the ynaF gene was determined by nucleotide sequencing and confirmed by comparison with the ynaF gene of S. typhimurium LT2 deposited in the EMBL data bank. The recombinant plasmid was transformed into BL21 (DE3) pLysS cells and plated on Luria–Bertani (LB) agar medium containing ampicillin. The pre-inoculum prepared from a single colony was transferred to a large culture of LB broth containing 50 µg ml−1 ampicillin and incubated at 310 K until the OD at 600 nm reached 0.6. For the preparation of soluble protein fractions, the cells from 1 l culture were pelleted, resuspended in 50 ml cold lysis buffer containing 50 mM Tris pH 8.0, 200 mM NaCl, 1% Triton X-100 and 10% glycerol and then lysed by sonication on ice. The clear supernatant containing soluble protein was collected by centrifugation.
2.2. Purification
YnaF with an N-terminal hexahistidine tag was purified using Ni–NTA His-Bind Resin (Novagen). All purification steps were performed at 277 K. All unbound proteins were washed from the column using 50 mM Tris–HCl pH 8.0, 200 mM NaCl (wash buffer). Nonspecifically bound proteins were washed from the column using wash buffer with 10–20 mM imidazole. YnaF protein was eluted from the column using wash buffer containing 200 mM imidazole. In order to remove the imidazole, the protein was extensively dialysed against 25 mM Tris–HCl pH 8.0, 2 mM mercaptoethanol. The dialysed protein was concentrated to 2.5 mg ml−1 using a 10 kDa molecular-weight cutoff Centricon (Amicon). 10 mg pure YnaF protein was obtained per litre of culture. The purified protein was examined using SDS–PAGE (Laemmli, 1970 ▶) and was found to be nearly homogenous (Fig. 1 ▶). The molecular weight and purity of the protein were checked using a MALDI–TOF mass spectrometer (Fig. 2 ▶).
Figure 1.
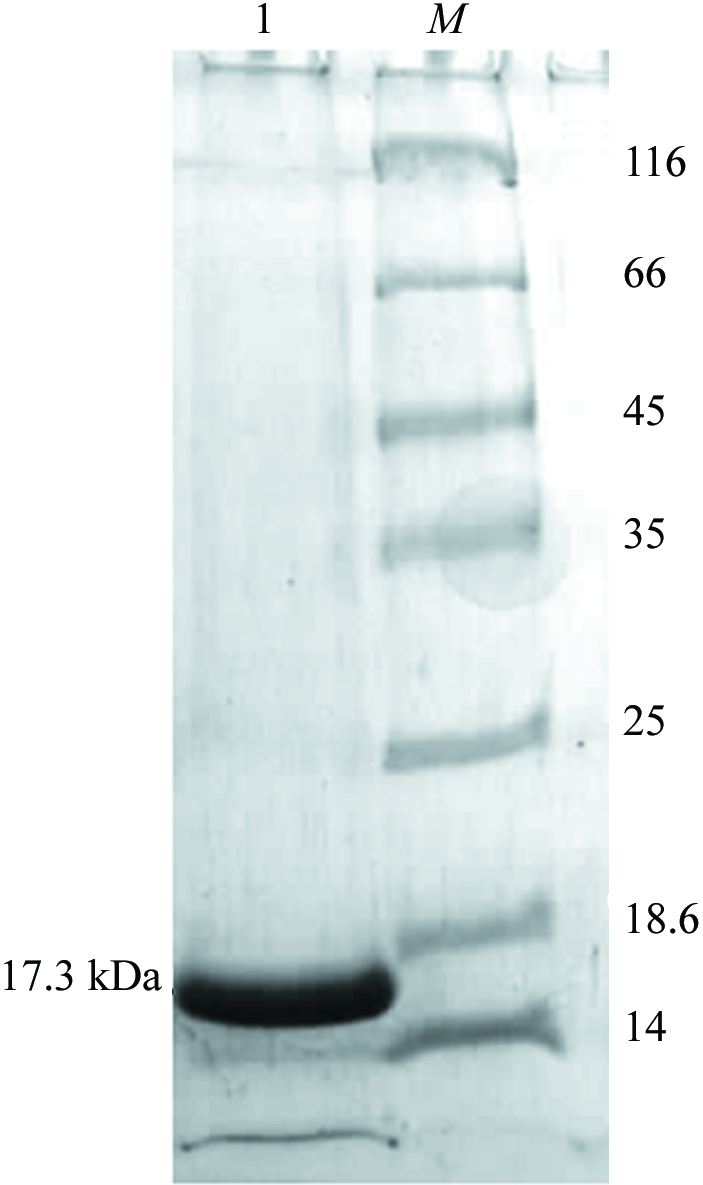
SDS–PAGE of YnaF. The protein was analyzed on 12% SDS–PAGE and stained with Coomassie blue. Lane 1, purified His-tagged YnaF; lane M, molecular-weight markers (Fermentas 431 Molecular-weight Marker Kit) labelled in kDa.
Figure 2.

Mass spectrum of YnaF obtained by MALDI–TOF. The peak corresponds to 17.3 kDa.
2.3. Crystallization
Initial crystallization experiments were carried out at 293 K with protein containing the hexahistidine tag and the hanging-drop vapour-diffusion and microbatch methods using Crystal Screen, Crystal Screen 2, PEG/Ion Screen, Index Screen and SaltRx Screen (Hampton Research). All crystallization drops were prepared by mixing 3 µl protein solution (2.5 mg ml−1) with 3 µl reservoir solution. In the hanging-drop method, the drop was suspended over 400 µl reservoir solution. In the initial screening, two hits were found: 1.6 M ammonium sulfate, 0.1 M MES pH 6.5, 10% dioxane (condition 1) and 10–20% PEG 3350, 0.2 M potassium iodide pH 6.8 (condition 2). Crystals of dimensions suitable for X-ray diffraction studies were obtained after 1–2 months (Fig. 3 ▶). Preliminary X-ray examination suggested that the crystals obtained under condition 1 diffracted to only 8 Å resolution, while those from condition 2 were suitable for structure determination.
Figure 3.

A single crystal of YnaF from S. typhimurium.
2.4. Data collection
Crystals were transferred to a cryoprotectant composed of reservoir solution (20% PEG 3350, 0.2 M KI) with 20% ethylene glycol for 30 s and then flash-cooled in liquid nitrogen. X-ray diffraction data were collected from a single crystal using a MAR Research image-plate system of diameter 345 mm. X-rays from a Rigaku RU200 rotating-anode X-ray generator equipped with a 300 µm focal cup were focused with Osmic mirrors. The crystal-to-detector distance was set to 200 mm. All frames were collected at 100 K with 0.75° oscillation angle and an exposure time of 900 s per frame. The data revealed significant spots to 2.5 Å resolution (Fig. 4 ▶). The data were indexed, integrated and scaled using DENZO and SCALEPACK from the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). Table 1 ▶ gives the data-collection statistics.
Figure 4.

Diffraction pattern obtained from a YnaF crystal.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 37.51, b = 77.18, c = 56.34, β = 101.8 |
| Resolution range (Å) | 30–2.5 (2.59–2.50) |
| Total No. of reflections | 15457 |
| No. of unique reflections | 10342 |
| Completeness (%) | 94.6 (86.7) |
| Rmerge† (%) | 12.5 (39.0) |
| 〈I〉/〈σ(I)〉 | 10.7 (2.4) |
| Matthews coefficient (Å3 Da−1) | 2.3 |
| No. of molecules per ASU | 2 |
R
merge = 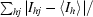
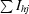 , where I
hj is the jth observation of reflection h and 〈I
h〉 is its mean intensity.
, where I
hj is the jth observation of reflection h and 〈I
h〉 is its mean intensity.
3. Results and discussion
Both the hanging-drop and microbatch methods gave crystals of comparable quality and of the same form using condition 2. A data set with 94.2% completeness was collected from a single crystal obtained using this condition and was processed to 2.5 Å resolution. Examination of systematic absences showed that the crystal belonged to space group P21. The volume of the asymmetric unit of the crystal was compatible with two protein subunits, with a volume per unit weight of 2.3 Å3 Da−1 and a calculated solvent content of 48% (Matthews, 1968 ▶). Molecular replacement using AMoRe (Navaza, 2001 ▶), Phaser (McCoy et al., 2005 ▶) and MOLREP (Vagin & Teplyakov, 2000 ▶) was carried out using homologous structures as search models (PDB codes 1mjh and 1wjg). These had sequence identities to the target sequence of 26 and 29%, respectively. No meaningful solution was found with any of these search models. Further structure-solution trials using multiple-wavelength anomalous dispersion are in progress.
Acknowledgments
The intensity data were collected at the X-ray facility for structural biology at the Molecular Biophysics Unit, Indian Institute of Science, supported by the Department of Science and Technology (DST) and the Department of Biotechnology (DBT) of the Government of India. SRS acknowledges the Council for Scientific and Industrial Research (CSIR), Government of India for the award of a fellowship.
References
- Gustavsson, N., Diez, A. & Nystrom, T. (2002). Mol. Microbiol.43, 107–117. [DOI] [PubMed]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Liu, W. T., Karavolos, M. H., Bulmer, D. M., Allaoui, A., Hormaeche, R. D., Lee, J. J. & Khan, C. M. (2007). Microb. Pathog.42, 2–10. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed]
- Nystrom, T. & Neidhardt, F. C. (1992). Mol. Microbiol.6, 3187–3198. [DOI] [PubMed]
- Nystrom, T. & Neidhardt, F. C. (1993). J. Bacteriol.175, 3949–3956. [DOI] [PMC free article] [PubMed]
- Nystrom, T. & Neidhardt, F. C. (1994). Mol. Microbiol.11, 537–544. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sambrook, J. & Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbour, NY, USA: Cold Spring Harbour Laboratory Press.
- Sousa, M. C. & McKay, D. B. (2001). Structure, 9, 1135–1141. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed]
- Walderhaug, M. O., Polarek, J. W., Voelkner, P., Daniel, J. M., Hesse, J. E., Altendorf, K. & Epstein, W. (1992). J. Bacteriol.174, 2152–2159. [DOI] [PMC free article] [PubMed]
- Zarembinski, T. I., Hung, L. W., Mueller-Dieckmann, H. J., Kim, K. K., Yokota, H., Kim, R. & Kim, S.-H. (1998). Proc. Natl Acad. Sci. USA, 95, 15189–15193. [DOI] [PMC free article] [PubMed]