

The X-ray crystal structure of the GTPase YjeQ from S. typhimurium is presented and compared with those of orthologues from T. maritima and B. subtilis.
Keywords: GTPase, YjeQ, ribosome
Abstract
The YjeQ class of P-loop GTPases assist in ribosome biogenesis and also bind to the 30S subunit of mature ribosomes. YjeQ ribosomal binding is GTP-dependent and thought to specifically direct protein synthesis, although the nature of the upstream signal causing this event in vivo is as yet unknown. The attenuating effect of YjeQ mutants on bacterial growth in Escherichia coli makes it a potential target for novel antimicrobial agents. In order to further explore the structure and function of YjeQ, the isolation, crystallization and structure determination of YjeQ from the enterobacterial species Salmonella typhimurium (StYjeQ) is reported. Whilst the overall StYjeQ fold is similar to those of the previously reported Thematoga maritima and Bacillus subtilis orthologues, particularly the GTPase domain, there are larger differences in the three OB folds. Although the zinc-finger secondary structure is conserved, significant sequence differences alter the nature of the external surface in each case and may reflect varying signalling pathways. Therefore, it may be easier to develop YjeQ-specific inhibitors that target the N- and C-terminal regions, disrupting the metabolic connectivity rather than the GTPase activity. The availability of coordinates for StYjeQ will provide a significantly improved basis for threading Gram-negative orthologue sequences and in silico compound-screening studies, with the potential for the development of species-selective drugs.
1. Introduction
YjeQ proteins form one of the five major subdivisions of the TRAFAC (translation-factor) family of P-loop GTPases and are broadly conserved in both Gram-positive and Gram-negative bacterial species (Fig. 1 ▶; Brown, 2005 ▶; Daigle et al., 2002 ▶). YjeQ from Escherichia coli (EcYjeQ) has been shown to possess slow GTPase activity (Daigle et al., 2002 ▶), which is stimulated 160-fold by interaction with the 30S component of the mature ribosome (Himeno et al., 2004 ▶; Daigle & Brown, 2004 ▶). Depletion of EcYjeQ or the Bacillus subtilis orthologue YloQ (BsYloQ) results in accumulation of 30S and 50S ribosomal subunits (Campbell et al., 2005 ▶; Himeno et al., 2004 ▶), suggesting a role in ribosome biogenesis; the B. subtilis mutant also exhibits filamentation (Campbell et al., 2005 ▶; Daigle & Brown, 2004 ▶). The recent construction and in vivo passage of yjeQ deletion mutants has shown that this gene is not essential, but nevertheless these strains have dramatically reduced growth rates, implying an important role in bacterial survival (Campbell et al., 2005 ▶; Himeno et al., 2004 ▶). This role of YjeQ, together with the absence of YjeQ/YloQ homologues in eukaryotes, may thus still make it a strong candidate target for the development of novel selective antimicrobial drugs.
Figure 1.

Amino-acid sequences for ten StYjeQ orthologues selected from those available via the IMG database. Alignment and visualization used the ClustalW/JALVIEW web-browser plugins, with colouring according to the ‘Zappo’ colour scheme (above 70% identity only). The top line of the figure indicates the various domains and loop regions (defined in the text). Rows 1–8 share similar genetic and sequence-organization patterns with StYjeQ, whilst rows 9 and 10 show the sequences of the T. maritima and B. subtilis orthologues, respectively.
Detailed biochemical data and ribosome-interaction studies have been reported for YjeQ from the enterobacterial species E. coli, for which no crystal structure is available. However, crystal structures have been determined for YjeQ orthologues from the anaerobic Gram-negative species Thermotoga maritima (TmYjeQ; Shin et al., 2004 ▶) and the aerobic Gram-positive species B. subtilis (BsYloQ; Levdikov et al., 2004 ▶). A similar protein fold is observed in both cases, with an N-terminal RNA oligonucleotide/oligosaccharide-binding domain, a central GTPase domain with ‘circularly permuted’ loop order (Shin et al., 2004 ▶), a zinc-finger domain and a short C-terminal extension. However, EcYjeQ shares less than 40% identity with these proteins; we therefore report the isolation, crystallization and structure determination of YjeQ from the enterobacterial species Salmonella typhimurium (StYjeQ), which should be highly relevant for the interpretation of the many biological studies available for the E. coli orthologue (96% amino-acid sequence identity). This work on StYjeQ forms part of a structural proteomics project aimed at characterizing a range of essential proteins as potential new targets for novel antibacterial drugs (Nichols et al., 2006 ▶, 2007 ▶).
2. Experimental methods
2.1. Isolation of recombinant StYjeQ
The S. typhimurium YjeQ-encoding DNA sequence was PCR-amplified and subcloned into the E. coli expression vector pET3a to yield the recombinant plasmid pMUT99. pMUT99 was transformed into E. coli strain BL21(DE3), which was grown at 303 K overnight in 500 ml volumes of Luria broth with selection by 100 µg ml−1 ampicillin. Induction took place with 0.2 mg ml−1 IPTG and cells were harvested by centrifugation after 5 h further incubation. Soluble protein was recovered by sonication and centrifugation, applied onto a Q-Sepharose column, washed with buffer A (50 mM potassium phosphate pH 7.2, 1 mM DTT) and eluted with a 0.0–1.0 M NaCl gradient. Pooled fractions were made 1.0 M with ammonium sulfate, applied onto a Phenyl Sepharose column in buffer A containing 1.0 M ammonium sulfate and eluted with a 1.0–0.0 M ammonium sulfate gradient in buffer A. Pooled fractions were dialysed twice with buffer A (2 × 5 l). The protein pool was loaded onto a hydroxyapatite column and eluted with a 0–400 mM potassium phosphate pH 7.2 gradient. Fractions containing StYjeQ were identified by SDS–PAGE at all column stages. This protocol yielded approximately 190 mg protein at greater than 98% purity from approximately 25 g cell paste. Selenomethionine-labelled (SeMet) StYjeQ, expressed in the B834 auxotroph strain, was also purified by the same procedure, yielding ∼150 mg pure protein from 5 g sonicated cell paste.
2.2. Dynamic light scattering
Dynamic light-scattering measurements on StYjeQ samples were carried out with a DynaPro-801 (Protein Solutions). Data were collected at room temperature from 1 mg ml−1 StYjeQ in buffer containing 0.1 M Hampton pH-screen solutions buffer pH 4–9, 0–500 mM NaCl with or without 2 mM GDP, GTP or GTP-γ-S.
2.3. Isothermal calorimetry (ITC)
ITC experiments were performed at 298 K using a high-precision VP-ITC system (Microcal Inc.). Protein was dialysed into 50 mM potassium phosphate pH 7.2, 1 mM β-mercaptoethanol and the dialysis buffer was used to dissolve the ligands GDP and GTP. The concentrations of the StYjeQ (cell) and the nucleotides (injector) are given in the legend to Table 1 ▶. The heat evolved following each 10 µl injection was obtained from the integral of the calorimetric signal. The heat arising from the binding reaction was obtained as the difference between the heat of reaction and the corresponding heat of dilution. Analysis of the data was performed using Origin software (Microcal ; Cooper, 1998 ▶; Cooper & Johnson, 1994 ▶).
Table 1. Thermodynamic parameters for the binding of GDP or GTP to StYjeQ as determined by ITC at 298 K.
The binding of GDP and GTP to StYjeQ was measured in triplicate in 50 mM potassium phosphate pH 7.2, 1 mM β-mercaptoethanol by ITC. The concentration of StYjeQ in the cell was 50 µM and the concentration of GDP or ATP in the injector was 690 µM. Shown are the values for n, the stoichiometry of binding, K d, the equilibrium dissociation constant, ΔH obs, the observed enthalpy, and ΔS0, the standard entropy change for single-site binding. The c values fall within the range 1–1000 that allows the isotherms to be accurately deconvoluted with reasonable confidence to derive K values (Wiseman et al., 1989 ▶).
| Titration 1 | Titration 2 | Titration 3 | Average | |
|---|---|---|---|---|
| GDP binding to StYjeQ | ||||
| n | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 |
| Ka | (1.4 ± 0.3) × 106 | (1.2 ± 0.2) × 106 | (0.7 ± 0.1) × 106 | (1.1 ± 0.3) × 106 |
| Kd,app (µM) | 0.7 | 0.8 | 1.4 | 1.0 ± 0.3 |
| ΔHobs (kJ mol−1) | −31.4 ± 0.8 | −31.4 ± 0.4 | −31.9 ± 0.8 | −31.4 ± 1.6 |
| ΔS0 (J K−1 mol−1) | 11.7 | 20.9 | 5.4 | 9.2 ± 3.3 |
| c | 67.5 | 60.1 | 38.5 | 55 ± 8.7 |
| GTP binding to StYjeQ | ||||
| n | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| Ka | (1.2 ± 0.1) × 105 | (1.1 ± 0.1) × 105 | (1.5 ± 0.1) × 105 | (1.3 ± 0.2) × 105 |
| Kd,app (µM) | 8.0 | 9.0 | 6.7 | 7.9 ± 0.8 |
| ΔHobs (kJ mol−1) | −39.3 ± 0.4 | −42.7 ± 0.8 | −38.9 ± 0.8 | −40.2 ± 2.1 |
| ΔS0 (J K−1 mol−1) | −38.9 | −47.3 | −31.0 | −39.3 ± 8.0 |
| c | 6.2 | 5.6 | 7.5 | 6.4 ± 1.0 |
2.4. Crystallization and data collection
Purified StYjeQ was buffer-exchanged into 10 mM Tris pH 7.4, 40 mM KCl using Vivascience Vivaspin 2 centrifugal concentrators, gel-filtered (Amersham NAP 25) and reconcentrated to 20 mg ml−1. A Cartesian robot system (Brown et al., 2003 ▶) was used to set up 100 + 100 nl sitting-drop crystallizations with 13 Hampton Research sparse-matrix and grid screens. Small crystals were observed in a single condition. Diffraction-quality crystals were obtained by optimizing conditions by hand in 6 µl droplets consisting of 3 µl well solution and 3 µl protein at 20 mg ml−1 equilibrated against 0.5 ml reservoir solution. The final conditions were 1.85 M ammonium sulfate, 0.1 M MES pH 6.5 and 10 mM cobalt(II) chloride. Crystals were flash-frozen in a 100 K nitrogen cold stream prior to data collection in-house using a MAR345 image-plate system on a Rigaku generator equipped with a Cu anode and Osmic multilayer optics to give Cu Kα radiation (λ = 1.5418 Å). Data images were indexed, integrated and merged using DENZO and SCALEPACK (Otwinowski & Minor, 1996 ▶; Otwinowski, 1993 ▶), indicating the space group to be P3121 or P3221; final statistics are shown in Table 2 ▶. Crystallization trials were also conducted with SeMet-labelled StYjeQ, including pH/PEG 3350 grid screens centred on the above optimized condition and seeding from droplets containing crystals of unlabelled StYjeQ. However, the SeMet-labelled material proved far more prone to precipitation and no crystals were obtained under any conditions tested.
Table 2. Data-collection and refinement statistics for S. typhimurium YjeQ.
Values in parentheses are for the outer shell.
| X-ray data statistics | |
| Space group | P3121 |
| Wavelength (Å) | 1.54179 |
| Unit-cell parameters (Å) | a = 92.84, c = 70.23 |
| Resolution range (Å) | 30.0–2.25 (2.30–2.25) |
| Refined mosaicity range (°) | 0.85–1.5 |
| Unique reflections | 16829 (1573) |
| Completeness (%) | 99.3 (94.1) |
| Redundancy | 6.1 (3.5) |
| Rmerge† | 0.104 (0.520) |
| 〈I/σ(I)〉 | 16.75 (1.99) |
| Wilson B factor (Å2) | 40.9 |
| Refined model statistics | |
| Protein residues in model | 270 |
| Water atoms | 248 |
| Ligand atoms | 30 |
| Rwork/Rfree‡ (%) | 19.9/26.8 |
| Ramachandran angles, most favoured (%) | 91.5 |
| Ramachandran angles, also allowed (%) | 8.5 |
| R.m.s.d. bond angles (°) | 1.23 |
| R.m.s.d. bond lengths (Å) | 0.005 |
| Mean B factors (Å2) | |
| Main chain | 36.5 |
| Side chain | 41.6 |
| Solvent | 45.8 |
R
merge = 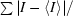
 , where I is the observed intensity of a given reflection and 〈I〉 is the averaged intensity for multiple measurements of this reflection.
, where I is the observed intensity of a given reflection and 〈I〉 is the averaged intensity for multiple measurements of this reflection.
R = 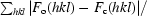
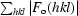 , where F
o are observed and F
c are calculated structure-factor amplitudes; the R
free set uses a randomly chosen 10% of reflections.
, where F
o are observed and F
c are calculated structure-factor amplitudes; the R
free set uses a randomly chosen 10% of reflections.
2.5. Structure determination
The StYjeQ crystal structure was readily solved by molecular replacement using the deposited coordinates of either TmYjeQ (PDB code 1u0l; Shin et al., 2004 ▶) or BsYloQ (PDB code 1t9h; Levdikov et al., 2004 ▶) as a model. The space group was assigned as P3121, with a single molecule in the asymmetric unit. However, the quality of phasing was rather poor and the resultant 2F o − F c and F o − F c electron-density maps were difficult to interpret. Further tests were therefore performed using Phaser (McCoy et al., 2005 ▶; Storoni et al., 2004 ▶) with both models defined as separate ensembles for the same component of the asymmetric unit, which greatly improved the accuracy of the starting phases. RESOLVE (Terwilliger, 2003 ▶, 2004 ▶) was then used to further reduce model bias by prime-and-switch phasing and also to build an initial model (∼72% of the expected number of residues, 44% sequence threaded) for further manual rebuilding with O (Jones et al., 1991 ▶). Refinement at each stage took place with CNS (Brünger et al., 1998 ▶), yielding a final model with R free = 26.2% and R work = 20.5%. Final statistics are shown in Table 2 ▶ and coordinates for the refined StYjeQ model have been deposited (PDB code 2rcn).
2.6. Model and sequence analysis
Model fitness was evaluated using PROCHECK (Laskowski et al., 1993 ▶). Pairwise Cα-trace overlays and r.m.s.d. calculations were generated with TOPP (Collaborative Computational Project, Number 4, 1994 ▶) and visualized in VMD (Humphrey et al., 1996 ▶), whilst multiple structural alignments were performed within VMD using the integrated alignment utility based on the STAMP algorithms (Eargle et al., 2006 ▶).
3. Results
High-purity recombinant StYjeQ, comprising 358 amino acids and with a molecular weight of 39.7 kDa, was isolated for this study using a series of hydrophobic interaction and affinity-chromatography purification steps. The yield was approximately 150 mg pure StYjeQ from 25 g cell paste for crystallization trials and biophysical testing (see §2).
3.1. Biophysical testing and calorimetry of StYjeQ
Dynamic light scattering was used to assess the oligomeric state of StYjeQ in aqueous solution at various pH values (pH 4–9) with variable amounts of NaCl (0–500 mM) and with or without 2 mM GDP, GTP or GTP-γ-S. Under all conditions tested, StYjeQ gave a single sharp peak in close agreement with the theoretical radius for monomeric proteins. These data suggest that StYjeQ forms a highly monodisperse monomer, a result which tallies with the previously reported 1:1 stoichiometry of interaction between EcYjeQ and the ribosome (Daigle & Brown, 2004 ▶). Dissociation constants have previously been determined for other P-loop GTPase classes (e.g. Era, with K d = 1.0 µM for GDP and K d = 5.5 µM for GTP; Chen et al., 1990 ▶), but equivalent values have not yet been reported for any YjeQ orthologues. Nucleotide binding by StYjeQ (GDP and GTP) was therefore assessed by isothermal titration calorimetry; typical results are shown in Figs. 2 ▶(a) and 2 ▶(b) and the associated thermodynamic parameters are summarized in Table 1 ▶. The data in Figs. 2 ▶(a) and 2 ▶(b) are in each case adequately described by a single-site binding model and Table 1 ▶ shows that StYjeQ bound both nucleotides with a 1:1 stoichiometry, with K d values of approximately 1 µM for GDP and 8 µM for GTP assessed in the absence of Mg2+ to prevent GTP turnover. These values are similar to those reported for Era, although in this case Mg2+ was present in the buffer (Chen et al., 1990 ▶).
Figure 2.

(a) and (b) Representative thermograms from isothermal calorimetric analysis of StYjeQ for (a) GTP binding and (b) GDP binding. (c) Representative electron density in the zinc-finger region of StYjeQ (chicken-wire representation of 2F obs − F calc density map contoured at 1σ, with waters shown as red spheres and the Zn2+ ion as a yellow sphere). The StYjeQ model is shown in stick format, with the thinner tracing indicating a side chain from a symmetry-related molecule. (d) Cartoon-format Cα trace of StYjeQ, illustrating domain structure and the location of the GDP- and Zn2+-binding sites. (e) 1.4 Å probe-radius SURF plot of the StYjeQ surface, showing the position of the bound GDP and illustrating the relationship between the binding cleft and the adjacent OB-fold domain. (f) Cartoon-format three-way alignment of YjeQ orthologues: StYjeQ (green), TmYjeQ (orange) and BsYloQ (purple) Cα traces.
3.2. Crystallization and structure determination
A wide range of nanoscale crystallization trials were set up covering unliganded StYjeQ as well as ligand cocrystallizations including GDP, GTP or GTP-γ-S. In general, trials with GTP-γ-S generated less precipitate, suggesting that its presence reduces nonspecific aggregation of StYjeQ. After four weeks crystals were observed in a single hit condition, which contained GTP-γ-S at setup. Equivalent trials with GDP and GTP did not generate crystals, but the refined structure clearly contains GDP not GTP-γ-S. These results therefore suggest that GDP binding induces conformational changes which not only allow StYjeQ to form the observed lattice, but also allow other modes of nonspecific binding. The GTP-γ-S added to the crystallization thus appears to act as a slow-release GDP source, helping to promote crystallization rather than aggregation. Representative electron density is shown in Fig. 2(c) ▶ and a cartoon-format Cα trace of the final model in shown in Fig. 2(d) ▶.
3.3. Fold description and comparison of StYjeQ with TmYjeQ and BsYloQ
StYjeQ is about 60 residues longer than TmYjeQ or BsYloQ. However, the majority of the additional residues are located at the N-terminus, forming an extra domain, with the remainder located in the C-terminal extension and the loop connecting the OB-fold to the GTPase domain; the remainder of the StYjeQ domain structure thus closely resembles those of TmYjeQ and BsYloQ (Fig. 1 ▶). As shown in Fig. 2(f) ▶, the overall StYjeQ structure is close to those of both TmYjeQ and BsYloQ, with greatest similarity in the zinc-finger and GTPase domains but more significant variation in the OB-fold and connecting loops (the overall mean Cα r.m.s.d. across all three structures is ∼1.8 Å for a 220-residue overlap). Pairwise comparisons using the SIM alignment algorithm show the largest overlap and highest BLAST score is with TmYjeQ versus BsYloQ rather than StYjeQ versus TmYjeQ; their corresponding three-dimensional structures also overlap more closely. It therefore seems that TmYjeQ may be more closely related to the B. subtilis protein than to StYjeQ. This relatedness may not be too surprising as although both T. maritima and S. typhimurium are Gram-negative species whilst B. subtilis is Gram-positive, sequence analysis shows a greater similarity between the genomes of B. subtilis and T. maritima (Nelson et al., 1999 ▶).
The additional N-terminal domain is disordered in our StYjeQ structure, but is designated as a probable ribosome-interaction domain (RID; residues 1–20; Fig. 1 ▶) on the basis of sequence homology with the equivalent region of EcYjeQ, in which it has been shown experimentally to play a critical role in the tight binding of EcYjeQ to the 30S ribosome (Daigle & Brown, 2004 ▶). Interestingly, sequence analysis reveals that a large group of YjeQ orthologues, including TmYjeQ and BsYloQ, do not possess this N-terminal domain, suggesting possible differences in the nature of their interaction with the ribosome. However, the larger form of YjeQ is not limited to Gram-negative species, as an orthologous cluster of Gram-positive species, including Mycobacterium tuberculosis, also possess similar N-terminal extensions, albeit with a sequence unrelated to that found in the gammaproteobacterial cluster.
The OB-fold binds ribosomal RNA and analysis of EcYjeQ mutants has shown that it is necessary for ribosome-induced stimulation of EcYjeQ GTPase activity (Daigle & Brown, 2004 ▶). However, the overall conservation of sequence between clusters of yjeQ orthologues is generally quite low in this region. Nonetheless, as shown in Fig. 2 ▶(f), the StYjeQ OB-fold secondary structure still closely matches those of TmYjeQ and BsYloQ, with two three-stranded antiparallel β-sheets packed orthogonally to one another, creating a β-barrel. The first β-strand of the N-terminal β-sheet is also shared with the second β-sheet, so the barrel has 1a↑2↓3↑4↑5↓1b↑ topology (Figs. 1 ▶, 2 ▶ d and 2 ▶ e; residues 39–101; OB-fold). This close structural homology most probably reflects the fact that the OB-fold tertiary structure is primarily determined by the optimal recognition determinants for RNA binding.
As with TmYjeQ and BsYloQ, the majority of the StYjeQ GTPase domain (Figs. 1 ▶, 2 ▶ d and 2 ▶ e; residues 121–271; GTPase) is formed by a six-stranded β-sheet with 3↑2↑1↑4↑6↑5↓ topology, giving rise to the loop order characteristic of ‘circularly permuted GTPases’ (Shin et al., 2004 ▶). Sequence analysis suggests that this domain is the most highly conserved between yjeQ orthologues and that the majority of residues that contact the bound GDP in our structure are also highly conserved amongst all YjeQ orthologues, although some equivalent substitutions are observed such as His201 in StYjeQ, which is equivalent to Lys149 in TmYjeQ.
The function of the C-terminal region encompassing the zinc-finger domain (residues 273–325; zinc-finger) and C-terminal extension (residues 326–358; CTE) is not yet known, but elsewhere the biological role of zinc-fingers is generally to either bind DNA/RNA or take part in protein–protein interactions. The zinc-coordinating contacts and secondary structure of the YjeQ zinc-finger are tightly conserved between StYjeQ, TmYjeQ and BsYloQ; sequence analysis also suggests that the same degree of conservation exists across all orthologues. However, larger differences are observed in the pattern of surface-exposed residues surrounding the zinc-finger and in the size and composition of the C-terminal extension that follows it.
4. Discussion
Comparison of the StYjeQ, TmYjeQ and BsYloQ structures shows a similar fold in all three cases, with the majority of conformational differences localizing to the loops of the OB-fold and the exposed surfaces of the C-terminal domain (Fig. 2 ▶ f). The overall conservation between three such widely separated bacterial species is striking, implying a strong link between structure and functional properties. Experimental data have shown that EcYjeQ assists in the biogenesis of ribosomes and also that it binds to the 30S subunit of the mature ribosome. In the latter case, ribosome association is stimulated 50-fold by the binding of GTP to EcYjeQ, which then induces a 160-fold increase in the rate of GTP hydrolysis. EcYjeQ deletion mutants also show increased susceptibility to antibiotics that target the A and P sites of the ribosome, preventing tRNA binding or inhibiting translation of the nascent protein chain (Campbell et al., 2005 ▶). These properties suggest that YjeQ plays a direct role in the regulation of protein synthesis and thus that it may function in a similar manner to other GTPases, providing a mechanical coupling that links the energy released by GTP hydrolysis to the energy required for protein synthesis. If this hypothesis is correct, one would expect YjeQ to be specifically activated in response to a signalling event. Furthermore, since the N-terminal and GTPase domains have known functions, it is possible that it is the C-terminal zinc-finger domain that interacts with one or more other partners to couple the biochemical activity of YjeQ to cellular metabolism.
Some support for this hypothesis can be gleaned from analysis of the genetic context and sequence conservation/variation between yjeQ orthologues. Such data show that the N- and C-terminal sequences cluster into groups, with strong conservation within a group. However, there is little conservation between groups, even where the GTPase domains remain much more closely related [sequences assessed using the IMG (Markowitz et al., 2006 ▶) and STRINGS (Snel et al., 2000 ▶) metaservers; data not shown]. Some species also have multiple paralogues, suggesting that YjeQ orthologues may have been recruited to different biochemical systems in various species. Additionally, sequence changes in the N-terminal regions are likely to reflect differences in the downstream signal propagation. Differing biochemical functions may thus be activated by YjeQ binding to the ribosome (in addition to GTP turnover). Similarly, changes in the C-terminal sequences are likely to reflect differences in the upstream components creating the signal that activates YjeQ in the first place.
YjeQ deletion strains are viable, but grow only very slowly (Campbell et al., 2005 ▶; Himeno et al., 2004 ▶); thus, YjeQ may prove viable as a novel target for drug development since microbial growth inhibition may lead to in vivo clearance. However, the GTP-binding cleft of YjeQ is structurally quite similar to those of many other GTP-binding proteins in both prokaryotes and eukaryotes. It may therefore be easier to develop YjeQ-specific inhibitors targeting the N- and C-terminal regions, disrupting the metabolic connectivity rather than the GTPase activity. In this case, inhibitors targeting these sites would be expected to be specific to a given species grouping owing to the high level of predicted structural variation. Such an approach would be predicted to improve the side-effect profile owing to higher specificity and to make any such inhibitors less prone to the selection and spread of drug resistance. Conversely, a narrower spectrum of antimicrobial activity would also reduce the commercial viability of such inhibitors. We therefore suggest that the development of YjeQ inhibitors as novel antimicrobial drugs would be best attempted only where fairly large groups of species show similarly conserved sequences. In this context, the largest single grouping we have identified is that which includes S. typhimurium, E. coli and the majority of other gammaproteobacterial species. It is thus hoped that the availability of the crystal structure of StYjeQ will be of use in providing a significantly improved starting model for threading these Gram-negative orthologue sequences and in silico compound-screening studies.
Supplementary Material
PDB reference: ribosomal interacting GTPase YjeQ, 2rcn, r2rcnsf
Acknowledgments
We thank J. Dong and Dr R. Esnouf for computer support, and Dr K. Harlos for help with X-ray data collection. We acknowledge the Biotechnology and Biological Sciences Research Council for funding this work through a LINK grant.
References
- Brown, E. D. (2005). Biochem. Cell Biol.83, 738–746. [DOI] [PubMed]
- Brown, J. et al. (2003). J. Appl. Cryst.36, 315–318.
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Campbell, T. L., Daigle, D. M. & Brown, E. D. (2005). Biochem. J.389, 843–852. [DOI] [PMC free article] [PubMed]
- Chen, S. M., Takiff, H. E., Barber, A. M., Dubois, G. C., Bardwell, J. C. & Court, D. L. (1990). J. Biol. Chem.265, 2888–2895. [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cooper, A. (1998). Methods Mol. Biol.88, 11–22. [DOI] [PubMed]
- Cooper, A. & Johnson, C. M. (1994). Methods Mol. Biol.22, 137–150. [DOI] [PubMed]
- Daigle, D. M. & Brown, E. D. (2004). J. Bacteriol.186, 1381–1387. [DOI] [PMC free article] [PubMed]
- Daigle, D. M., Rossi, L., Berghuis, A. M., Aravind, L., Koonin, E. V. & Brown, E. D. (2002). Biochemistry, 41, 11109–11117. [DOI] [PubMed]
- Eargle, J., Wright, D. & Luthey-Schulten, Z. (2006). Bioinformatics, 22, 504–506. [DOI] [PubMed]
- Himeno, H., Hanawa-Suetsugu, K., Kimura, T., Takagi, K., Sugiyama, W., Shirata, S., Mikami, T., Odagiri, F., Osanai, Y., Watanabe, D., Goto, S., Kalachnyuk, L., Ushida, C. & Muto, A. (2004). Nucleic Acids Res.32, 5303–5309. [DOI] [PMC free article] [PubMed]
- Humphrey, W., Dalke, A. & Schulten, K. (1996). J. Mol. Graph.14, 33–38. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Levdikov, V. M., Blagova, E. V., Brannigan, J. A., Cladiere, L., Antson, A. A., Isupov, M. N., Seror, S. J. & Wilkinson, A. J. (2004). J. Mol. Biol.340, 767–782. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Markowitz, V. M., Ivanova, N., Palaniappan, K., Szeto, E., Korzeniewski, F., Lykidis, A., Anderson, I., Mavromatis, K., Kunin, V., Garcia Martin, H., Dubchak, I., Hugenholtz, P. & Kyrpides, N. C. (2006). Bioinformatics, 22, e359–e367. [DOI] [PubMed]
- Nelson, K. E. et al. (1999). Nature (London), 399, 323–329.
- Nichols, C. E., Johnson, C., Lockyer, M., Charles, I. G., Lamb, H. K., Hawkins, A. R. & Stammers, D. K. (2006). Proteins, 64, 111–123. [DOI] [PubMed]
- Nichols, C. E., Lamb, H. K., Lockyer, M., Charles, I. G., Pyne, S., Hawkins, A. R. & Stammers, D. K. (2007). Proteins, 68, 13–25. [DOI] [PubMed]
- Otwinowski, Z. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 56–62. Warrington: Daresbury Laboratory.
- Otwinowski, Z. & Minor, W. (1996). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Shin, D. H., Lou, Y., Jancarik, J., Yokota, H., Kim, R. & Kim, S.-H. (2004). Proc. Natl Acad. Sci. USA, 101, 13198–13203. [DOI] [PMC free article] [PubMed]
- Snel, B., Lehmann, G., Bork, P. & Huynen, M. A. (2000). Nucleic Acids Res.28, 3442–3444. [DOI] [PMC free article] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Terwilliger, T. (2004). J. Synchrotron Rad.11, 49–52. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol.374, 22–37. [DOI] [PubMed]
- Wiseman, T., Williston, S., Brandts, J. F. & Lin, L. N. (1989). Anal. Biochem.179, 131–137. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ribosomal interacting GTPase YjeQ, 2rcn, r2rcnsf