

Abstract
Numerous studies have established that polyvalency is a critical feature of cell surface carbohydrate recognition. Nevertheless, carbohydrate–protein interactions are typically evaluated by using assays that focus on the behavior of monovalent carbohydrate ligands in solution. It has generally been assumed that the relative affinities of monovalent carbohydrate ligands in solution correlate with their polyvalent avidities. In this paper we show that carbohydrate ligands synthesized directly on TentaGel beads interact with carbohydrate-binding proteins in a polyvalent manner. The carbohydrate-derivatized beads can, therefore, be used as model systems for cell surfaces to evaluate polyvalent carbohydrate–protein interactions. By using a combinatorial approach to synthesize solid-phase libraries of polyvalent carbohydrates, one can rapidly address key issues in the area of cell surface carbohydrate recognition. For example, studies reported herein demonstrate that there is an unanticipated degree of specificity in recognition processes involving polyvalent carbohydrates. However, the correlation between polyvalent avidities and solution affinities is poor. Apparently, the presentation of carbohydrates on the polymer surface has a profound influence on the interaction of the ligand with the protein receptor. These findings have implications for how carbohydrates function as recognition signals in nature, as well as for how polyvalent carbohydrate–protein interactions should be studied.
Keywords: combinatorial chemistry, carbohydrate recognition, cell adhesion, parallel screening, lectin
Interactions between carbohydrates on the surface of one cell type and proteins on the surface of another cell type play critical roles in a wide variety of biochemical recognition processes (1, 2). However, the details of these interactions are poorly understood. Typically, receptor–ligand binding events are studied by making derivatives of the ligand and directly quantitating the affinity. Applying this approach to cell surface carbohydrates has been problematic because carbohydrates are notoriously difficult to synthesize; it is usually not feasible to make more than a small number of derivatives, and even that can take years (3). Moreover, it is difficult to measure the binding affinities by using direct methods because individual carbohydrates bind weakly (Kd ≈ 10−3 M) to their protein receptors. Therefore, the relative affinities of carbohydrates are obtained from the concentrations of ligand required to inhibit some event or process—e.g., cell agglutination—that is caused by interactions between the protein receptor and carbohydrates presented on the cell surface. These inhibition assays have shown that many carbohydrate-binding proteins can bind a variety of different structures with similar affinities (4). The broad specificity makes it hard to evaluate which structural features are critical for recognition.
Despite the low affinity and broad specificity of individual carbohydrate–protein interactions, carbohydrates function as very specific signals in a wide variety of cell–cell recognition events. Proteins involved in carbohydrate recognition typically have multiple binding sites. Since the carbohydrate ligands are usually presented in clusters on the cell surface, these proteins can bind more than one carbohydrate simultaneously (5, 6). Studies on model systems indicate that, although the affinities of monovalent carbohydrates for their protein receptors are weak, a polyvalent display of carbohydrates produces high avidities (7–16). Recent evidence also indicates that specificity increases when carbohydrates are presented in a polyvalent format (17–19). It has been proposed that the “intrinsic” affinities of monovalent carbohydrate ligands are amplified by a polyvalent presentation (19).
It is critical to establish whether polyvalency amplifies the intrinsic affinities of monovalent carbohydrate ligands because the result has implicatons for how carbohydrate–protein interactions are evaluated. If polyvalency does amplify the intrinsic affinities of carbohydrate ligands, then studies on the binding in solution of monovalent carbohydrate ligands should provide valid information about the relative avidities of the corresponding polyvalent ligands. Moreover, it would be reasonable to design synthetic polyvalent carbohydrate ligands based on information about monovalent affinities. However, there is little experimental evidence to support the hypothesis that the solution affinities of monovalent carbohydrates correlate well with their polyvalent avidities. Earlier we reported a strategy to synthesize large numbers of polyvalent carbohydrate ligands on TentaGel beads (20). Below we show that these carbohydrate-derivatized beads behave in key respects as model systems for cell surfaces. The carbohydrate-derivatized beads can, therefore, be used to study important issues in polyvalent carbohydrate–protein recognition processes. We have found that there is not a good correlation between the solution affinities of carbohydrate ligands and their polyvalent avidities, presumably because the presentation of carbohydrates on surfaces has a profound effect on their interactions with various receptors. Moreover, there is a remarkable degree of specificity in polyvalent carbohydrate–protein recognition. The implications of these findings are discussed below.
MATERIALS AND METHODS
Materials.
TentaGel resin (TentaGel S NH2, 130 μm, 0.3 mmol⋅g−1 capacity) was purchased from Rapp Polymere (Tübingen, Germany). Dichloromethane (CH2Cl2), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), diisopropylethylamine (DIEA), and trifluoroacetic acid (TFA), and bovine serum albumin (BSA) were from Aldrich. 1-Methyl-2-pyrrolidinone (NMP), 1-hydroxybenzotriazole (HOBt) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were from Applied Biosystems. Lyophilized powders of lectins and alkaline phosphatase-conjugated streptavidin, as well as solutions of 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitroblue tetrazolium (NBT) liquid substrate system and p-nitrophenyl phosphate were from Sigma. Rabbit blood was purchased from Remel (Lenexa, KS). Phosphate-buffered saline (PBS) was 150 mM NaCl/7.3 mM Na2HPO4/2.7 mM NaH2PO4, adjusted to pH 7.2. PBST is 0.05% Tween-20 in PBS. Tris-buffered saline (TBS) was 500 mM NaCl/20 mM Tris, adjusted with dilute HCl to pH 7.5. TBST was 0.05% Tween-20 in TBS. Alkaline phosphatase buffer was 100 mM NaCl/5 mM MgCl2/100 mM Tris, adjusted to pH 9.5 with HCl.
General Procedure for the Synthesis of Resin-Bound Disaccharides.
TentaGel resin (0.674 g) was suspended in 15 ml of NMP, and to this was added 4′-(carboxylic acid)methyleneoxyphenyl 3-O-acetyl-2-azido-4,6-O-benzylidine-2-deoxy-1-thio-α-d-galactopyranoside (0.122 g, 0.243 mmol), DIEA (0.22 g, 1.3 mmol), and HOBt/HBTU solution (0.45 M in DMF, 2.2 g, 0.93 mmol). The suspension was shaken for 12 h, and the resin was washed with CH2Cl2, NMP, and DMF. A solution of anhydrous hydrazine in DMF (1:7, vol/vol; 16 ml) was added, and the reaction mixture was shaken for 9 h until acetate hydrolysis was shown to be complete by infrared analysis (potassium bromide pellet). The resin was washed with DMF, H2O, methanol, and CH2Cl2 and encoded for the glycosyl acceptor (21, 22). A resin portion (0.100 g) was suspended in 5 ml of CH2Cl2 and agitated with argon for 10 min. Phenyl 2,3,4,6-tetra-O-pivaloyl-1-thio-β-d-galactopyranoside sulfoxide (0.24 g, 0.40 mmol) and 2,6-di-tert-butyl-4-methylpyridine (0.13 g, 0.65 mmol) were dissolved in anhydrous CH2Cl2 (5 ml) and added to the resin. The suspension was cooled to −60°C and a solution of trifluoromethanesulfonic anhydride (34 μl, 0.20 mmol) in 1 ml of CH2Cl2 was added. After warming to 0°C over 1–2 h, the resin was washed successively with saturated sodium bicarbonate, H2O, methanol, diethyl ether, CH2Cl2, and toluene. The resin was dried and resubjected to the glycosylation conditions. The resin was encoded, suspended in thiolacetic acid (25 ml), shaken at room temperature for 27 h, washed with CH2Cl2, and dried. The resin was suspended in 20% tetrahydrofuran (THF)/CH2Cl2 (20 ml), shaken at room temperature for 30 min, and washed with CH2Cl2. The resin was allowed to swell in a solution of 20% THF/MeOH (20 ml) for 10 min, and ground lithium hydroxide monohydrate (0.20 g, 4.8 mmol) was added. The reaction mixture was shaken at room temperature for 11 h and washed with H2O until the pH of the filtrate was determined to be neutral. The resin was then dried in vacuo for 12 h.
Aggregation Study.
One-milligram samples of TentaGel beads derivatized with Galβ1,3GalNAcβ-thiophenyl glycoside and underivatized beads were placed in separate wells of a 96-well microtiter plate and swollen in PBS. The buffer was removed and 100 μl of Arachis hypogaea lectin (23) (10–1000 μg/ml in PBS) was added to each well. The beads were examined under a microscope after 1–2 h.
Hemagglutination Assay.
A stock solution of the lectin was made by dissolving 5 mg of Bauhinia purpurea lectin (24, 25) in 2.5 ml of PBS. Serial dilutions of lectin in PBS were prepared, and 50 μl of each solution was transferred into 12 microtiter plate wells. To each well 50 μl of a 2% suspension of rabbit erythrocytes in PBS was added, and the plate was incubated at room temperature on an orbital shaker for 1 h. Agglutinated cells formed a carpet covering the bottom of the well; nonagglutinated cells formed a compact button at the center of the well. The titer was defined as the last dilution well before the erythrocyte button begins to form. The hemagglutination assay titer for Bauhinia purpurea lectin was 1 μg/ml.
Hemagglutination Inhibition Assay.
Stock solutions (5 mg/ml) of 1b–4b (see Table 1) in PBS were prepared and serial dilutions were made. Twenty-five microliters of each sugar solution was added to single wells of a microtiter plate that contained 25 μl of a 16 μg/ml lectin solution in PBS and incubated at room temperature on an orbital shaker for 1 h. To each well was added 50 μl of a 2% suspension of erythrocytes. The plate was gently agitated for 10 min and then incubated at room temperature for 1 h. The final lectin concentration was 4 μg/ml, which was 4 doses of the hemagglutination assay titer. The end point was defined as the lowest sugar concentration that inhibited agglutination.
Table 1.
Results of the screening of the four-carbohydrate mixture

Represents concentration of ligand needed to inhibit agglutination of erythrocytes. All values were based on ligand 1b taken as 1.0.
a series R = OCH2C(O)NH-TentaGel.
b series R = H.
c series R = OCH2C(O)NHCH2CH2CH2OCH3.
**ND = not determined.
Colorimetric Assay for Four-Carbohydrate Mixture.
A portion of resin that contained equal portions of 1a–4a (10 mg total) was washed with PBST (three times with 1 ml for 10 min). The beads were incubated for 30 min at room temperature in 1 ml of PBST containing 3% BSA and washed with PBST (three times with 1 ml for 5 min) containing 1% BSA. The beads were incubated in 1 ml of Bauhinia purpurea lectin (0.1 μg/ml in PBST containing 1% BSA) at room temperature for 3 h and then washed with TBST (3 times with 1 ml for 5 min) containing 1% BSA. The resin was incubated for 20 min at room temperature in 1 ml of alkaline phosphatase-conjugated streptavidin (10 μg/ml in TBST containing 1% BSA) and then washed with alkaline phosphatase buffer (three times with 1 ml for 5 min). A small portion of the resin (≈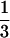 ) was stained with 200 μl of BCIP/NBT. Staining was terminated after 30 min by washing the beads twice with 200 μl of 20 mM sodium ethylenediaminetetraacetic acid, pH 7.4. The dark purple, light purple, and colorless beads were pulled out for decoding by using 50-μl micropipettes.
) was stained with 200 μl of BCIP/NBT. Staining was terminated after 30 min by washing the beads twice with 200 μl of 20 mM sodium ethylenediaminetetraacetic acid, pH 7.4. The dark purple, light purple, and colorless beads were pulled out for decoding by using 50-μl micropipettes.
Colorimetric Assay for Carbohydrate Library.
Screening of the carbohydrate library followed the same procedure as in the four-compound mixture except the lectin concentration was 10 μg/ml for the library (20).
Inhibition Assay.
TentaGel resin (1.0 mg) derivatized with Galβ1,3GalNAcβ-thiophenyl (1a) was added to each well of a 96-well MultiScreen Filtration Plate (Millipore). Each portion of resin was washed with PBST (three times with 100 μl for 5 min). The buffer solution was removed from each well simultaneously by placing the filtration plate on a MultiScreen Vacuum Manifold (Millipore). Each portion of resin was incubated for 30 min with 100 μl of PBST containing 3% BSA and washed with PBST containing 1% BSA (three times with 100 μl for 5 min). Sugar solutions of 10 different concentrations were prepared from sugar stock solutions (5 mg/ml in PBST containing 1% BSA). A solution of Bauhinia purpurea lectin (1 mg/ml in PBS) was added to each well to afford a final lectin concentration of 100 μg/ml. The combined lectin/sugar solutions were incubated at room temperature for 1 h, and 100 μl of each solution was added to the resin. The plate was agitated on an orbital shaker at room temperature for 3 h. The resin was washed with TBST containing 1% BSA (three times with 100 μl for 5 min). A solution (100 μl) of alkaline phosphatase-conjugated streptavidin (10 μg/ml in TBST containing 1% BSA) was added to each well, and the plate was incubated at room temperature for 20 min. The beads were washed with alkaline phosphatase buffer (three times with 100 μl for 5 min) and transferred into a 96-well flat-bottomed microtiter plate. A p-nitrophenyl phosphate solution (100 μl) was added to each well with a 12-channel Pipetman (Brinkmann) and the color development at 405 nm was monitored using a microplate reader.
RESULTS AND DISCUSSION
To date, most polyvalent carbohydrate model systems have been constructed by attaching previously synthesized carbohydrates to some type of scaffold. Our ultimate goal was to produce large numbers of polyvalent carbohydrate ligands simultaneously in a format that would permit parallel screening. By synthesizing carbohydrates directly on a solid support and screening them for binding, we hoped to exploit some of the advantages of combinatorial chemistry (20, 26, 27) in studying carbohydrate recognition processes (see below).
Selecting the Solid Support.
Two criteria were important in choosing a solid support: ease of synthesis and ease of screening. We had previously developed methods to synthesize carbohydrates on the Merrifield polystyrene resin (28). However, control experiments established that the Merrifield beads aggregate in water and that proteins adsorb nonspecifically to the polystyrene surface. Whitesides and co-workers (29, 30) have shown that nonspecific protein adsorption to gold surfaces can be minimized by derivatizing the surfaces with polyethylene glycol. Others have established that TentaGel, a polyethylene glycol-derivatized polystyrene resin which swells in both organic solvents and aqueous buffers, is well suited for displaying ligands, including carbohydrates, for biological screening (31). To evaluate the suitability of the TentaGel resin for synthesizing and screening carbohydrate libraries, the disaccharide Galβ1,3GalNAc (1a) (Fig. 1) was constructed on TentaGel by using the chemistry previously developed for the Merrifield resin. All of the chemical transformations, including the glycosylation reaction, worked extremely well in terms of both stereochemical outcome and yield.
Figure 1.

Synthesis of the resin-bound disaccharide Galβ1,3GalNAcβSPh. T, TentaGel resin. Conditions were as follows: a, HOBt/HBTU, DIEA, NMP, room temperature, 12 h; b, NH2NH2/DMF (1:7), room temperature, 9 h; c, trifluoromethanesulfonic anhydride, DTBMP, CH2Cl2, −60°C to 0°C; repeat; d, thiolacetic acid, room temperature, 27 h; e, 20% trifluoroacetic acid/CH2Cl2, room temperature, 30 min; and f, LiOH, 80% MeOH/tetrahydrofuran, room temperature, 11 h.
The synthesis was deliberately carried out from the reducing to the nonreducing end of the ligand so that the carbohydrate ligand would be presented on the TentaGel bead in a way that mimics the presentation of cell surface carbohydrates. We hoped this would allow us to directly screen carbohydrate-derivatized beads for binding to protein receptors (32). To evaluate the potential of the TentaGel resin for on-bead screening, we treated separate samples of the Galβ1,3GalNAc-derivatized beads as well as underivatized beads with various concentrations of Arachis hypogaea (peanut) lectin (23), a protein known to recognize Galβ1,3GalNAc. The carbohydrate-derivatized beads aggregated at a lectin concentration of 25 μg/ml (Fig. 2). In contrast, the underivatized beads did not aggregate even at lectin concentrations as high as 1,000 μg/ml. It should be noted that Arachis hypogaea lectin causes erythrocytes to aggregate—i.e., agglutinate—at low concentrations in a process believed to involve polyvalent interactions between the lectin and carbohydrates on the surfaces of different cells. Thus, the behavior of the derivatized TentaGel beads in the presence of the lectin suggests that the derivatized beads aggregate due to multivalent interactions between the lectin and carbohydrate ligands on different beads. Consistent with this hypothesis, aggregation can be prevented by increasing the protein concentration significantly. Under these conditions, there is sufficient protein to coat the entire surface of each bead, making multivalent interactions involving carbohydrates on different beads impossible. Thus, with respect to multivalent recognition, the derivatized beads appear to mimic the behavior of cell surface carbohydrates, suggesting that they may serve as good model systems for studying cell surface carbohydrate–protein recognition events.
Figure 2.

Aggregation study of TentaGel beads. (A) Underivatized beads did not aggregate when treated with Arachis hypogaea lectin at 25 μg/ml. (B) Derivatized beads aggregated under the same conditions. (×100.)
Development of a Colorimetric Assay for Detecting Binding.
Although the initial investigations on the resin-bound carbohydrates with the lectin were promising, an aggregation assay cannot be used to screen mixtures of polyvalent carbohydrate ligands. We required an assay that could potentially discriminate between different carbohydrate-derivatized beads. Assays in which receptor binding is detected by a visible change in the appearance of individual beads are well suited for parallel screens. However, given the broad recognition specificity of protein receptors for monovalent carbohydrate ligands, it was important to determine whether it would be possible to distinguish between closely related polyvalent carbohydrate ligands in a parallel screen.
To address this question, four similar carbohydrate ligands (1a–4a) were synthesized on TentaGel resin (Table 1). Galβ1,3GalNAc (1a) is a known ligand for Bauhinia purpurea lectin (24, 25). The structures of the three other carbohydrates differed from 1a in terms of the stereochemistry at the C4 position and/or at the anomeric position of the sugar directly attached to the resin. The C4 stereochemistry was varied because solution binding studies have shown that the lectin is sensitive to the stereochemistry at this position and binds to the C4-equatorial isomer with an affinity 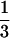 of that for the axial isomer (24, 25). The configuration of the internal glycosidic linkage was varied to probe the effect of ligand presentation on binding (33, 34).
of that for the axial isomer (24, 25). The configuration of the internal glycosidic linkage was varied to probe the effect of ligand presentation on binding (33, 34).
The beads were encoded (21, 22) during the synthesis so that the four carbohydrates could be screened in parallel and the results assessed quickly. To screen the beads, a sample of resin containing equal amounts of the four different carbohydrate-derivatized beads was incubated with biotinylated Bauhinia purpurea lectin (0.1 μg/ml), followed by streptavidin-linked alkaline phosphatase. The beads were then stained with BCIP/NBT, which is converted by alkaline phosphatase to an insoluble purple polymer that precipitates on the surface of the beads. Beads that stain darkly presumably have more enzyme-linked conjugate, and hence more bound lectin, than the other beads. Approximately 25% of the beads stained very darkly, 25% of the beads stained lightly, and 50% of the beads did not stain (Fig. 3). Twenty dark purple beads, 20 light purple beads, and 18 unstained beads were removed from the mixture and decoded to determine the identity of the carbohydrates. All 20 dark purple beads contained Galβ1,3GalNAcβ-thiophenyl (1a), but none of the light purple or unstained beads contained this ligand (Table 1). Thus, the assay clearly distinguishes the best ligand from three other closely related ligands. It was also apparent from the assay that the worst polyvalent ligand is 4a, in which both the C4 and the anomeric stereochemistry differ from the known ligand, 1a. Ligands 2a and 3a have similar avidities, although the ratio of stained to unstained beads suggests that 2b is a better ligand. Hence, the two best ligands contained the β-stereochemistry at the internal glycosidic linkage.
Figure 3.

Colorimetric assay of the four-carbohydrate mixture. (×100.)
To evaluate the relationship between the polyvalent avidities and monovalent affinities of the carbohydrate ligands, we synthesized the thiophenyl glycosides 1b–4b and evaluated their relative solution affinities for Bauhinia purpurea lectin using a standard hemagglutination inhibition assay (35). The results showed that 1b inhibits agglutination at concentrations ¼ to 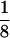 of those for the other three thiophenyl glycosides. There was essentially no difference in the solution affinities of the other three carbohydrates. Furthermore, changing from the thiophenyl glycosides to a set of thiophenyl derivatives containing an acetamide ethanolamine chain (1c–4c), which resembles the linker to the resin, had no effect on the relative solution affinities. Hence, the acetamide ethanolamine chain does not appear to interact with the protein.
of those for the other three thiophenyl glycosides. There was essentially no difference in the solution affinities of the other three carbohydrates. Furthermore, changing from the thiophenyl glycosides to a set of thiophenyl derivatives containing an acetamide ethanolamine chain (1c–4c), which resembles the linker to the resin, had no effect on the relative solution affinities. Hence, the acetamide ethanolamine chain does not appear to interact with the protein.
The above results suggest that there is a poor correlation between solution affinities and on-bead avidities. The on-bead screen shows detectable differences between the polyvalent avidities of the four carbohydrate ligands. In contrast, the agglutination inhibition assay shows that only one of the four carbohydrates has a measurably higher binding affinity. Although the best inhibitor in solution proved to be the best polyvalent carbohydrate ligand, further work has suggested that this correlation does not hold up when larger numbers of compounds are studied (see below). In this regard, it should be noted that previous investigations of the relationship between solution affinities and polyvalent avidities have involved small numbers of different carbohydrate compounds.
Screening of a Carbohydrate Library.
Having developed an assay for carbohydrate binding that can discriminate between a small number of closely related ligands, we were ready to evaluate the utility of a combinatorial approach for studying carbohydrate recognition. Although the parallel screen gave unambiguous results in selecting the best of four compounds, it was by no means clear that it would be possible to screen a library containing hundreds or thousands of related carbohydrates.
An encoded library designed to contain ≈1,300 different compounds with 72 different glycosidic linkages was synthesized (20). Using an assay procedure similar to that described above for the four-compound mixture, we screened 10 mg of the resin-bound library, or approximately six copies of each carbohydrate, against Bauhinia purpurea lectin at a concentration of 10 μg/ml. Fewer than 0.3% of the beads in a pool of ≈9,000 beads stained significantly over a period of 20 min. Twenty-five dark purple beads were selected from the library over a period of 20 min and decoded (Table 2). Five copies each of two closely related compounds 5a and 6a were identified among the 25 beads. Both compounds contained the same disaccharide structure, Galα1,3GlcN-acyl-α-thiophenyl with a hydrophobic N-acyl group (4-nitrobenzoyl or isovaleryl). Three other beads contained the same disaccharide structure with different, but also hydrophobic, N-acyl groups.
Table 2.
Results from the screening of the carbohydrate library

In the upper part of the table, 13 beads that contained the same core disaccharide were found in the library. In the lower part, 12 beads found in the library were considered to be noise in the colorimetric assay. (All of the compounds belong to the a series.)
Beads that stained dark purple were pulled out from the library after 5-, 10, and 20-min staining periods.
a series R = OCH2C(O)NH-TentaGel; b series R = H; and c series R = OCH2C(O)NHCH2CH2OCH3.
The assay was repeated three times with similar results. In all cases, only a small number of beads stained, and multiple copies of compounds 5a and 6a were selected from the library. Of the remaining stained beads, no pattern was evident either within individual runs or between runs. These beads presumably represent the noise in the enzyme-linked assay. It should be noted that 5a and 6a were selected only when the beads were incubated with the lectin; they did not show up in control assays in which the beads were incubated with streptavidin-alkaline phosphatase alone. Thus, the library screen showed that it is possible to obtain a remarkable degree of specificity in carbohydrate recognition.
Although the results were unambiguous, the structures of the preferred ligands 5a and 6a surprised us. These ligands contain an α-glycosidic linkage between the two sugars, whereas the known ligand for this lectin contains a β linkage. Changing the stereochemistry of the linkage between the two sugars changes the overall shape of the molecule significantly. In addition, the preferred ligands have an equatorial hydroxyl group at the C4 position of the resin-linked sugar even though the results from screening the small library had suggested that the axial hydroxyl group is preferred. Finally, the preferred ligands have an axial anomeric linkage to the resin. The screen of the four-compound mixture had suggested that the equatorial thiophenyl linkage to the resin is preferred over the axial stereochemistry, at least when the glycosidic linkage between the two sugars is equatorial. Although both the α- and β-thiophenyl derivatives of the known ligand, Galβ1,3GalNAc, were included in the library, neither appeared among the pool of stained beads.
To verify the results of the screen, we separately synthesized on TentaGel the two best ligands, 5a and 6a, as well as the known ligand, 1a. Portions of beads containing the three compounds were then mixed and screened. The two ligands 5a and 6a stained rapidly, while the known ligand, 1a, did not stain in their presence. However, control experiments again showed that the known ligand stained, albeit at higher lectin concentrations, in the presence of beads containing other, less preferred, carbohydrate derivatives. Thus, the ligands identified from the library bind the lectin more avidly than the known ligand, despite containing a number of structural changes that are individually unfavorable.
To evaluate the relationship between monovalent binding affinities in solution and polyvalent avidities, 1c, 5c, and 6c were synthesized and assayed for their ability to inhibit lectin binding to TentaGel beads derivatized with Galβ1,3GalNAc, 1a. All compounds inhibited lectin binding in the micromolar range, consistent with the broad recognition specificity that carbohydrate-binding proteins typically display for monovalent carbohydrates. However, the known ligand 1c inhibited binding at 25 μM and was considered to be the best monovalent ligand in solution. 5c and 6c inhibited lectin binding at 35 and 46 μM, respectively. It is clear that the solution affinities of monovalent ligands do not correlate well with the corresponding polyvalent avidities. We have concluded that the presentation of the carbohydrates on the polymer beads has a profound influence on their avidities. We would expect that the presentation of carbohydrates on cell surfaces also influences their receptor-binding interactions. Therefore, it is important to exercise caution in drawing conclusions about the structure–function relationships of surface-bound carbohydrates from solution affinities.
Conclusion.
Although polyvalent carbohydrate recognition events are often studied using monovalent ligands to probe structure–function relationships, our results demonstrate that there is not a good correlation between solution affinities and polyvalent avidities. Thus, it is critical to have readily accessible polyvalent model systems. The results presented above show that carbohydrates synthesized directly on polymer beads bind in a polyvalent manner to carbohydrate-binding proteins. These carbohydrate-derivatized beads can, therefore, serve as polyvalent model systems to probe structure–function relationships in carbohydrate binding and to identify new polyvalent carbohydrate ligands for various receptors.
Another approach to studying polyvalent carbohydrate interactions was recently reported by Bertozzi and co-workers (36), who showed that carbohydrates can be attached to bioengineered cell surfaces by means of a hydrazone linker. This clever approach permits one to evaluate carbohydrate–receptor interactions in a context that contains other, potentially relevant, cell surface components. Our approach is complimentary to Bertozzi’s because it permits one to study carbohydrate–receptor interactions in a context that is isolated from other interactions. An isolated system is critical to answer many questions about carbohydrate–protein interactions. For example, there has been considerable debate about the role of carbohydrates in cellular recognition processes. Because the differences in the affinity between carbohydrate ligands in solution are small, it has often been assumed that cell surface carbohydrates are relatively nonspecific binding elements. The paradox, of course, is that carbohydrates are involved in many exquisitely specific biological events. In some cases, the specificity has been attributed to secondary protein–protein interactions, which are established only after an initial nonspecific carbohydrate-mediated adhesion process. Our results from the parallel screen of the ≈1,300-compound carbohydrate library have definitively established that carbohydrate–protein interactions can be remarkably specific; there are no other components present that could be responsible for the observed specificity.
One issue that our work has not adequately addressed is whether the observed specificity is a result of kinetic factors or thermodynamics. Our results show that the avidity of surface-bound ligands and their solution affinities do not correlate. We do not know whether this is because attaching ligands to a surface alters their “intrinsic affinities” (a thermodynamic effect) for a particular receptor, or whether it simply affects the on-rates of different ligands. Because the off-rates of polyvalent carbohydrate–protein interactions are extremely slow, it is possible that the specificity observed both in our screen and in nature is due to kinetic factors. To address mechanistic aspects of the specificity, it is critical to evaluate on- and off-rates of various ligands in more carefully defined systems, varying ligand density and linker presentation.
Acknowledgments
This work was supported by the Office of Naval Research and a James A. Shannon Director’s Award from the National Institutes of Health (D.K.). J.L.L. was supported by the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation (DRG-1365).
ABBREVIATIONS
- DMF
N,N-dimethylformamide
- DIEA
diisopropylethylamine
- NMP
1-methyl-2-pyrrolidinone
- HOBt
1-hydroxybenzotriazole
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- BCIP
5-bromo-4-chloro-3-indolyl phosphate
- NBT
nitroblue tetrazolium
- Gal
galactose
- Glc
glucose
- Man
mannose
- Fuc
fucose
References
- 1.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharon N, Lis H. Sci Am. 1993;268(1):82–89. doi: 10.1038/scientificamerican0193-82. [DOI] [PubMed] [Google Scholar]
- 3.Toshima K, Tatsuta K. Chem Rev. 1993;93:1503–1531. [Google Scholar]
- 4.Drickamer K. Struct Biol. 1995;2:437–439. doi: 10.1038/nsb0695-437. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y C. FASEB J. 1992;6:3193–3200. doi: 10.1096/fasebj.6.13.1397841. [DOI] [PubMed] [Google Scholar]
- 6.Bundle D R, Young N M. Curr Opin Struct Biol. 1992;2:666–673. [Google Scholar]
- 7.Kiessling L L, Pohl N L. Chem Biol. 1996;3:71–77. doi: 10.1016/s1074-5521(96)90280-x. [DOI] [PubMed] [Google Scholar]
- 8.Sigal G, Mammen M, Dahmann G, Whitesides G. J Am Chem Soc. 1996;118:3789–3800. [Google Scholar]
- 9.Adler P, Wood S J, Lee Y C, Lee R T, Petri J, W A, Schnaar R L. J Biol Chem. 1995;270:5164–5171. doi: 10.1074/jbc.270.10.5164. [DOI] [PubMed] [Google Scholar]
- 10.Sabesan S, Duus J O, Domaille P, Kelm S, Paulson J C. J Am Chem Soc. 1991;113:5865–5866. [Google Scholar]
- 11.Sabesan S, Duus J, Neira S, Domaille P, Kelm S, Paulson J C, Bock K. J Am Chem Soc. 1992;114:8363–8375. [Google Scholar]
- 12.DeFrees S, Phillips L, Guo L, Zalipsky S. J Am Chem Soc. 1996;118:6101–6104. [Google Scholar]
- 13.Spengard U, Schudok M, Wolfgang S, Kretzschmar G, Kunz H. Angew Chem Int Ed Engl. 1996;35:321–324. [Google Scholar]
- 14.Roy R. Curr Opin Struct Biol. 1996;6:692–702. doi: 10.1016/s0959-440x(96)80037-6. [DOI] [PubMed] [Google Scholar]
- 15.Meunier S, Roy R. Tetrahedron Lett. 1996;37:5469–5472. [Google Scholar]
- 16.Welply J, Abbas S, Scudder P, Keene J, Broschat K, Casnocha S, Gorka C, Steininger C, Howard S, Schmuke J, Graneto M, Rotsaert J, Manger I, Jacob G. Glycobiology. 1994;4:259–265. doi: 10.1093/glycob/4.3.259. [DOI] [PubMed] [Google Scholar]
- 17.Lees W, Spaltenstein A, Kingery-Wood J, Whitesides G. J Med Chem. 1994;37:3419–3433. doi: 10.1021/jm00046a027. [DOI] [PubMed] [Google Scholar]
- 18.Weatherman R V, Mortell K H, Chervenak M, Kiessling L L, Toone E J. Biochemistry. 1996;35:3619–3624. doi: 10.1021/bi951916z. [DOI] [PubMed] [Google Scholar]
- 19.Mortell K, Weatherman R, Kiessling L. J Am Chem Soc. 1996;118:2297–2298. [Google Scholar]
- 20.Liang R, Yan L, Loebach J, Ge M, Uozumi Y, Sekanina K, Horan N, Gildersleeve J, Thompson C, Smith A, Biswas K, Still W C, Kahne D. Science. 1996;274:1520–1522. doi: 10.1126/science.274.5292.1520. [DOI] [PubMed] [Google Scholar]
- 21.Nestler P, Bartlett P, Still W C. J Org Chem. 1994;59:4723–4724. [Google Scholar]
- 22.Ohlmeyer M H J, Swanson R N, Dillard L W, Reader J C, Asouline G, Kobayashi R, Wigler M, Still W C. Proc Natl Acad Sci USA. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lotan R, Skutelsky E, Danon D, Sharon N. J Biol Chem. 1975;250:8518–8523. [PubMed] [Google Scholar]
- 24.Osawa T, Irimura T, Kawaguchi T. Methods Enzymol. 1978;50:367–372. doi: 10.1016/0076-6879(78)50044-x. [DOI] [PubMed] [Google Scholar]
- 25.Wu A M, Kabat E A, Gruezo F G, Allen H J. Arch Biochem Biophys. 1980;204:623–639. doi: 10.1016/0003-9861(80)90074-0. [DOI] [PubMed] [Google Scholar]
- 26.Gallop M, Barrett R, Dower W, Fodor S, Gordon E. J Med Chem. 1994;37:1233–1251. doi: 10.1021/jm00035a001. [DOI] [PubMed] [Google Scholar]
- 27.Gordon E, Barrett R, Dower W, Fodor S, Gallop M. J Med Chem. 1994;37:1385–1401. doi: 10.1021/jm00036a001. [DOI] [PubMed] [Google Scholar]
- 28.Yan L, Taylor C M, Goodnow R, Jr, Kahne D. J Am Chem Soc. 1994;116:6953–6954. [Google Scholar]
- 29.Prime K L, Whitesides G M. Science. 1991;252:1164–1167. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- 30.Prime K L, Whitesides G M. J Am Chem Soc. 1993;115:10714–10721. [Google Scholar]
- 31.Bayer E. Angew Chem Int Ed Engl. 1991;30:113–129. [Google Scholar]
- 32.Vetter M, Tate E M, Gallop M. Bioconj Chem. 1995;6:319–322. doi: 10.1021/bc00033a014. [DOI] [PubMed] [Google Scholar]
- 33.Glick G D, Knowles J R. J Am Chem Soc. 1991;113:4701–4703. [Google Scholar]
- 34.Glick G D, Toogood P L, Wiley D C, Skehel J J, Knowles J R. J Biol Chem. 1991;266:23660–23669. [PubMed] [Google Scholar]
- 35.Rogers G N, Pritchett T J, Lane J L, Paulson J C. Virology. 1983;131:394–408. doi: 10.1016/0042-6822(83)90507-x. [DOI] [PubMed] [Google Scholar]
- 36.Mahal L K, Yarema K J, Bertozzi C R. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]