

Abstract
Cockayne syndrome (CS) is characterized by impaired physical and mental development. Two complementation groups, CSA and CSB, have been identified. Here we report that the CSB gene product enhances elongation by RNA polymerase II. CSB stimulated the rate of elongation on an undamaged template by a factor of about 3. A thymine-thymine cyclobutane dimer located in the template strand is known to be a strong block to transcription. Addition of CSB to the blocked polymerase resulted in addition of one nucleotide to the nascent transcript. Finally, addition of transcription factor IIS is known to cause polymerase blocked at a thymine-thymine cyclobutane dimer to digest its nascent transcript, and CSB counteracted this transcript shortening action of transcription factor IIS. Thus a deficiency in transcription elongation may contribute to the CS phenotype.
Keywords: transcription
Cockayne syndrome (CS) is characterized by abnormal and slow development that becomes evident within the first few years after birth. “Cachetic dwarfism” describes the outward appearance of afflicted individuals. Patients are mentally retarded due to impaired neurological development. Some patients are photosensitive, but this photosensitivity has not been associated with increased incidence of skin cancer. Also commonly present are cataracts, retinal pigmentary degeneration, dental caries, and hearing loss. The mean age of death is 12 years (1, 2).
The characteristics of CS at the cellular level include lack of transcription-repair coupling (TRC) and abnormally slow recovery of RNA and DNA synthesis following UV-irradiation (3, 4). At the biochemical level, TRC is a process in which damage in the template strand of an active gene undergoes nucleotide excision repair faster than damage in the coding strand or nontranscribed DNA (5, 6). Thus in CS cells, damage in nontranscribed DNA is repaired at the same rate as in normal cells. Deficient repair in CS cells is limited to the template strand of the small fraction of the genome that is actually transcribed.
The mechanisms behind these responses of CS cells to UV are not well defined, although complementation analyses suggest that at least two proteins, CSA and CSB are involved (7, 8). Based upon its sequence, the CSA protein, which is 44 kDa in size, is considered a member of the WD-repeat family of proteins (9). This family is known to have regulatory but not enzymatic activity (10). The function of CSA protein is otherwise unknown. The CSB protein has been considered a possible human TRC factor based on several structural and functional similarities with the TRC factor in Escherichia coli, the Mfd protein (11). Similar to Mfd, CSB is rather large with a molecular mass of 168 kDa (12). Both Mfd and CSB possess a “helicase motifs” region (11, 12), ATPase and DNA binding activities, but lack helicase activity (12, 13). Mfd brings about TRC by removing a stalled RNA polymerase from the DNA template and delivering repair enzymes to the damage site (11). In contrast, CSB does not remove a stalled RNA polymerase II (RNAPII) from the DNA template (13). Thus if CSB is a TRC factor, then the coupling mechanism in humans is likely to be different than that in E. coli.
In fact, an in vitro study utilizing reconstituted human transcription and nucleotide excision repair systems found that coupling occurs in the absence of CSB protein (14). In that study, elongating RNAPII was blocked by a thymine-thymine cyclobutane dimer (T<>T) in the template. Even though the blocked RNAPII was very stable, repair of the transcription-blocking lesion from the ternary complex was as efficient as repair of a T<>T in naked DNA. In contrast, repair of damage in nucleosomal DNA is inhibited compared with repair of naked DNA (15, 16). Thus in vivo, RNAPII stalled at a T<>T may make the T<>T more accessible to repair enzymes than a lesion in a nucleosome.
Consequently, CSB may have no direct role in TRC. Since it has been suggested that CS might arise from defective transcription (2, 17–19), and CSB protein has recently become available for biochemical analysis, we have tested the effect of CSB on transcription. We find that in fact CSB enhances transcription elongation by RNAPII. This enhancement may indirectly stimulate TRC and indicates that a deficiency in transcription elongation may contribute to the clinical manifestations of CS.
MATERIALS AND METHODS
Materials.
The plasmid pMLU112 that contains the adenovirus major late promoter has been described (20). The first 112 nucleotides of the coding strand downstream of the transcription start site contains no T residues and is referred to as a “U-less cassette.” Hence, transcription in the absence of UTP ends with a transcript of 112 nucleotides. pPU192, described previously (14), contains the major late promoter and a T<>T located in the template strand at nucleotides 149–150 downstream from the transcription start site.
Methods.
We utilized a reconstituted transcription system (13, 14) consisting of purified recombinant human TFIIB, IIE, IIF, native human IIH and RNAPII, and recombinant yeast TATA box binding protein. Enzymes (≈1.2 ng TATA box binding protein/0.4 ng IIB/8.0 ng IIE/0.8 ng IIF/2.9 ng IIH/0.001 units RNAPII; units as in ref. 21) were incubated with 2 ng of pMLU112 in 3.3 μl consisting of 60 mM Hepes (pH 7.9), 6 mM Tris (pH 7.9), 108 mM KCl, 6.4 mM MgCl2, 2.1 mM EDTA, 4 mM DTT, 2.8 mM 2-mercaptoethanol, 5% glycerol, and 3% polyethylene glycol. The RNAPII used for transcription was largely in the RNAPIIA form while the preparation used in binding assays (see below) was largely in the RNAPIIO form. Both forms were isolated from HeLa cells. After 30 min preincubation at 28°C, 125 μM ATP, 125 μM GTP, and 6 μM [α-32P]CTP were added. Incubation continued for 45 min. Reactions were then diluted with 2 volumes of a solution to arrive at 8.7 mM Tris (pH 7.9), 30 mM Hepes (pH 7.9), 61 mM KCl, 13 mM NaCl, 5.4 mM MgCl2, 0.9 mM 2-mercaptoethanol, 5% glycerol, 1% polyethylene glycol, 1.7 mM ATP, 42 μM GTP, 2 μM CTP 133 μg/ml BSA, and 8 units of RNasin (Promega). Incubations continued with 150 ng of purified CSB (13) and 10 ng of purified TFIIS (a gift of D. Reinberg, University of Medicine and Dentistry, Piscataway, NJ) as indicated. Reactions were then stopped by adding 6 mM EDTA, 0.12% SDS, 60 mM sodium acetate (pH 5.5), and 0.6 mg/ml tRNA, and mixing with and equal volume of phenol/chloroform/isoamyl alcohol. Following organic extraction, the products were resolved on 6% sequencing gels. Nucleotides were not removed in experiments with TFIIS. For the elongation assay, 15 ng of CSB was added, and reactions were chased by adding UTP to 400 μM and unlabeled CTP to 800 μM. pPU192 was transcribed as described for pMLU112 except UTP (125 μM) was included.
To end label transcripts for RNA sequencing analysis with RNase T1, pPU192 was transcribed first with ATP and [α-32P]CTP, which are the only nucleotides incorporated in the first ten nucleotides of the RNA, and then transcripts were chased by adding UTP, GTP, and excess cold CTP.
The pull-down assay utilized a construct containing residues 528-1222 of CSB fused to maltose binding protein. This construct and assay methods were described (13).
RESULTS
CSB Stimulates Transcriptional Elongation.
To investigate the effect of CSB on transcription by RNAPII we employed a reconstituted transcription system and the template pMLU112, which allows formation of a 112-nucleotide transcript containing no U residues. CSB was incubated with RNAPII, the general transcription factors, pMLU112, ATP, GTP, and 32P-CTP, and then the amount of 112-nucleotide RNA product was measured We found that after a 6 min incubation, the presence of CSB increased the amount of 112-nucleotide RNA product by 2.2-fold compared with a control transcription reaction, and after 60 min CSB increased the amount of product by 1.3-fold (not shown).
Numerous steps are required for formation of the 112-nucleotide transcript. We first examined the possibility that CSB enhances the elongation step. When RNAPII stalls after forming the 112-nucleotide transcript, it forms a stable complex. CSB or storage/dilution buffer was added to the stable ternary complexes, and then transcription was chased by adding UTP plus cold CTP to dilute the radiolabel. Transcripts formed after 1–3 min elongation are shown in Fig. 1A, lanes 2–5. Clearly CSB enhanced the rate of elongation by almost 3-fold. This stimulation of elongation most likely accounts for the stimulation of formation of the 112-nucleotide product seen in preliminary experiments. The stimulation of elongation is caused by CSB because removal of CSB from the reaction by anti-CSB antibodies completely abolished the stimulatory effect of the CSB preparation (Fig. 1B). CSB also stimulated transcription past the attenuation site located in the adenovirus major late transcriptional unit (ref. 22; data not shown). Finally, CSB enhanced elongation when transcription was performed using a promoterless (dC-tailed) template (23). Addition of general transcription factors are not required for transcription of this template, and stimulation of elongation by CSB was observed independently of added general transcription factors (data not shown).
Figure 1.

CSB stimulates transcription elongation. RNAPII was stalled at the end of the 112-nucleotide long U-less cassette in pMLU112 by omission of UTP. (A) CSB was added to 9 nM (lanes 3 and 5), then reactions were chased by adding UTP, and CTP was added to dilute the radiolabel. After 0–3 min of chasing, reactions were stopped and resolved on a sequencing gel alongside DNA markers (lane M) of the sizes indicated. The effect of CSB was optimal when it was present at 6–20 nM. (B) The CSB preparation was immunoprecipitated in the presence and absence of anti-CSB antibody. Gel analysis (not shown) indicated that the antibody did in fact precipitate CSB protein. The supernatants were added to transcription reactions with CSB nominally at 10 nM, and transcription was chased as in A for 1 min
Interaction of CSB with RNAPII.
CSB is known to bind to DNA and thus the enhancement of elongation could be considered a secondary effect of a CSB-DNA interaction. However, the enhancement of elongation was seen at subnanomolar concentrations of CSB (not shown), a condition in which CSB would not efficiently bind to DNA (13). Therefore we tested whether CSB could interact with RNAPII. A pull-down assay was used, in which a partial construct of CSB was fused to maltose binding protein (MBP). The MBP-CSB fusion protein, “MBPE65,” bound to amylose resin, was incubated with RNAPII. Most of the polymerase was present in the phosphorylated form (RNAPIIO) present in elongation complexes. The results in Fig. 2 show that both unphosphorylated and phosphorylated forms of RNA polymerase bound quantitatively to MBPE65, with no background binding to MBP. Therefore it is likely that CSB stimulates elongation as a result of an interaction with RNAPII in elongation complex.
Figure 2.
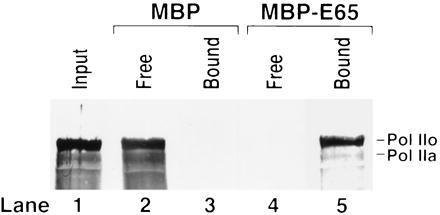
CSB-RNAPII interaction. A pull-down assay utilized MBP-CSB, a fusion protein of amino acids 528-1222 of CSB fused to the C terminus of MBP (13). MBP and the MBP-CSB fusion protein, “MBPE65” each attached to amylose at 1 g/l, were incubated with purified RNAPII. Resins were pelleted and unbound (Free) proteins in the supernatants were removed. Resins with “Bound” proteins were washed. Components of Free and Bound fractions were separated on a SDS/polyacrylamide gel and analyzed by Western blot using monoclonal anti-RNAPII C-terminal domain antibodies (Promega). Both phosphorylated (RNAPIIo) and nonphosphorylated (RNAPIIa) forms of RNAPII bound to CSB.
Effect of CSB on RNAPII Stalled by DNA Damage.
Elongation by RNAPII is known to be completely blocked by a T<>T in the template strand (24). To examine a possible role of CSB on the blocked polymerase we constructed pPU192, which possesses a T<>T located in the template strand at positions 149–150 downstream from the adenovirus major late promoter transcription start site (14). We then mapped the 3′ end of the RNA made when RNA polymerase is blocked by the T<>T. Transcription of pPU192 resulted in several products, as shown in the gels in Fig. 3A (lane 1), and Fig. 3B (lane 2), and represented schematically in Fig. 3C. The major product, labeled “a”, is 148 nt long and ends 1 nucleotide before the T<>T. Minor products “b” and “c” are 149 nucleotides and 150 nucleotides, respectively, and end across from the T<>T. The 146-nucleotide band “P” is considered a pause site since it was not formed when undamaged DNA was transcribed (Fig. 3B, lane 1), but it was formed in response to a downstream impediment to transcription, either the T<>T, (lane 2), or the end of the template after position 156, (lane 4).
Figure 3.
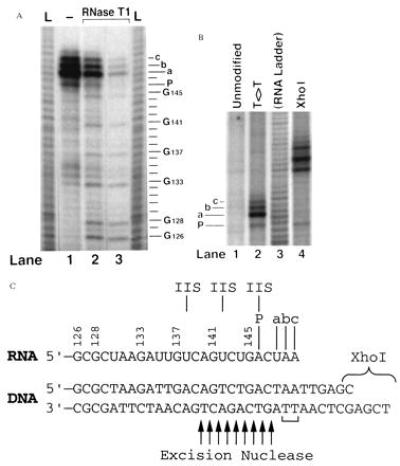
Transcription termination site in relation to the T<>T. In Panel A, end labeled transcripts terminated at the T<>T were digested 3′ to G residues using 1/100,000 (lane 2) and 1/30,000 (lane 3) dilutions of RNase T1 (1931 units/μl, GIBCO/BRL). Products were resolved alongside undigested RNA (lane 1) and RNA ladders (L) generated by alkaline hydrolysis. Lines to the right indicate RNA residues. G residues and termination sites a, b, c, and P are labeled. (B) Transcripts made from unmodified pPU192, pPU192 digested with XhoI, or pPU192 with a T<>T in the template strand. (C) DNA and RNA sequences surrounding the T<>T (indicated with a bracket). Termination sites a, b, c, and P and three major cleavage products obtained in the presence of TFIIS are labeled above the RNA. Arrows below indicate where the excision repair nuclease nicks the damaged strand 3′ to the lesion (25). The RNA sequence opposite the dimer may vary from that shown due to the damage.
CSB added to RNAPII stalled at the T<>T did not promote elongation past the lesion (not shown). However, as shown in Fig. 4A, lanes 1 and 2, CSB did promote elongation as indicated by the addition of one nucleotide to the major transcript, band a, converting it to band b. In the absence of CSB, the ratio of transcripts a/b was 2.31 ± 0.60, and in the presence of CSB, the ratio was 0.75 ± 0.33, based upon scans of gels such as in Fig. 4A. This means about 3-fold stimulation by CSB of the activity of RNAPII to insert a nucleotide across from the 3′ thymine of the thymine dimer. In addition, CSB enhanced transcription past the pause site P.
Figure 4.
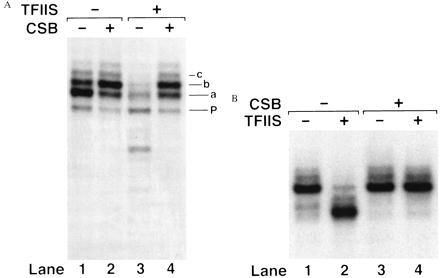
Effects of CSB plus TFIIS on blocked RNAPII. (A) Stalled elongation complexes were formed at the T<>T located in the template strand of pPU192. Reactions were then incubated with CSB and TFIIS for 25 min at 30°C. Transcripts a, b, c, and P are indicated. The experiment shown in B utilized RNAPII stalled at the end of the 112-nucleotide U-less cassette in pMLU112. (B) CSB and TFIIS were incubated with the stalled complex for 25 min at 30°C.
CSB Does Not Promote TFIIS-Induced Transcript Shortening.
It has been proposed that TFIIS acts as a TRC factor (25–27). TFIIS causes RNAPII stalled at a T<>T to digest its nascent transcript by several nucleotides (24). It was suggested that this transcript shortening, likely aided by CSB, might in some way expose of the transcription-blocking lesion to repair enzymes. Therefore we tested whether CSB in fact facilitates transcript shortening.
We observed the expected transcript shortening when TFIIS alone was added to RNAPII stalled at a T<>T (Fig. 4A, lane 3; see also schematic in Fig. 3C). Transcripts were shortened by 2, 6, and 10 nucleotides with additional bands in the 3–13 range appearing at lower frequencies. In contrast to predictions of TRC models (25–27), CSB counteracted the effect of TFIIS (Fig. 4A, lane 4). Reduced levels of CSB had no effect on the transcript shortening action of TFIIS (not shown). CSB also counteracted the effect of TFIIS on RNAPII stalled by nucleotide starvation at the end of the 112-nucleotide U-less cassette (Fig. 4B). Thus the anti-IIS effect of CSB is not restricted to a site of damage, and in a sense constitutes a third way in which CSB enhances elongation by RNAPII.
DISCUSSION
The principal finding of this investigation is that CSB enhances elongation by RNAPII. The enhancement of transcription is most likely the consequence of an interaction of CSB with elongating RNAPII. The mechanism of CSB is closer to that of TFIIF and ELL than to TFIIS (and its prokaryotic counterparts GreA and GreB), which induce degradation of the nascent transcript at pause sites (28, 29). Our finding has implications in the pathogenesis of CS and in the mechanism of TRC.
Regarding the pathogenesis of CS, the hallmark of CS cells in culture is their unique response to DNA damage, i.e., lack of TRC and slow recovery of RNA synthesis (3, 4). However, these aberrant responses to DNA damage have not been established as causes for CS (19). An alternative hypothesis is that CS arises from a deficiency in transcription. This idea arose to explain the fact that CS and related disorders can also be caused by mutations in components of transcription factor IIH, specifically the subunit encoded by the Xeroderma Pigmentosum group D gene (17, 18). This transcription-CS link is complicated by the fact that TFIIH is also required for nucleotide excision repair (30). However, recently it was reported that CSB cells and permeabilized CSB cells exhibit a deficiency in transcription. This deficiency was ascribed to either an altered chromatin organization in CSB cells or an inefficient transcription complex organization (31). Our findings provide direct biochemical evidence that at face value support the hypothesis that a deficiency in transcription in some way contributes to the development of CS.
Our experiments fail to support the notion that TFIIS functions as a TRC factor in conjunction with CSB. Of relevance, Verhage and coworkers (32) found that disruption of the gene encoding TFIIS had no effect on TRC in yeast.
To date, CSB has been found to have no direct effect on transcription-dependent or -independent nucleotide excision repair (unpublished observation). However, it has been found that a T<>T in a ternary complex can be readily repaired by the human excision repair system (14). Since RNAPII stalled at damage may make the damage more accessible to repair enzymes than damage within a nucleosome, this may be considered a form of TRC that is independent of CSB. A mechanism such as this may also constitute the coupling factor (rad 26)-independent pathway of TRC identified in yeast (33). The data presented in this report suggest three indirect roles for CSB in TRC. First, by stimulating elongation, CSB may increase the rate at which polymerase becomes blocked at lesions and elicits repair. This could involve nucleotide excision repair and possibly other forms of repair such as base excision repair (34). A second possible role of CSB in coupled repair may be to increase the effective concentration of repair factors at the lesion site. CSB can bind to XPA, TFIIH (13) and XPG (32) and is shown here to interact with RNAPII. Finally, it is possible that TFIIS inhibits repair where polymerase is stalled. Nucleotide excision repair is initiated with formation of a nick in the damaged strand 2–10 phosphodiester bonds 3′ to the lesion (25), as illustrated in Fig. 3C. TFIIS thus causes degradation of the transcript directly across from the 3′ incision site; repeated shortening and elongation of the transcript at this site may inhibit repair. CSB, however, positions and stabilizes the polymerase at the site of the dimer.
Further research on improving the efficiencies of the in vitro systems for transcription by RNAPII and repair by human excision nuclease and biochemical characterization of CSA are needed for a better understanding of the stimulation of repair by transcription in humans.
Acknowledgments
This work was supported by National Institutes of Health Grant GM32833 and by the Council for Tobacco Research, Inc., Grant CTR3852.
ABBREVIATIONS
- CS
Cockayne syndrome
- RNAPII
RNA polymerase II
- TRC
transcription-repair coupling
- T<>T
thymine-thymine cyclobutane dimer
- MBP
maltose binding protein
References
- 1.Nance M A, Berry S A. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg E C, Walker G C, Siede W. DNA Repair Mechanisms. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 3.Mayne L V, Lehmann A R. Cancer Res. 1982;42:1473–1478. [PubMed] [Google Scholar]
- 4.Venema J, Mullenders L H, Natarajan A T, van Zeeland A A, Mayne L V. Proc Natl Acad Sci USA. 1990;87:4707–4711. doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohr V A, Smith C A, Okumoto D S, Hanawalt P C. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 6.Mellon I, Spivak G, Hanawalt P C. Cell. 1985;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka K, Kawai K, Kamahara Y, Ikenaga M, Okada Y. Somatic Cell Genet. 1981;7:445–455. doi: 10.1007/BF01542989. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann A R. Mutat Res. 1982;106:347–356. doi: 10.1016/0027-5107(82)90115-4. [DOI] [PubMed] [Google Scholar]
- 9.Henning K A, Li L, Iyer N, McDaniel L D, Reagan M S, Legerski R, Schultz R A, Stefanini M, Lehmann A R, Mayne L V, Friedberg E C. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 10.Neer E J, Schmidt C J, Nambudripad R, Smith T F. Nature (London) 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 11.Selby C P, Sancar Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 12.Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers J H J. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 13.Selby C P, Sancar A. J Biol Chem. 1997;272:1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- 14.Selby C P, Drapkin R D, Reinberg D R, Sancar A. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Xiaohua W, Friedberg E C. J Biol Chem. 1991;266:22472–22478. [PubMed] [Google Scholar]
- 16.Sugasawa K, Masutani C, Hanaoka F. J Biol Chem. 1993;268:9098–9104. [PubMed] [Google Scholar]
- 17.Bootsma D, Hoeijmakers J H J. Nature (London) 1993;363:114–115. doi: 10.1038/363114a0. [DOI] [PubMed] [Google Scholar]
- 18.Vermeulen W, van Vuuren A J, Chipoulet M, Schaeffer L, Appeldoorn E, Weeda G, Jaspers N G J, Priestley A, Arlett C F, Lehmann A R, Stefanini M, Mezzina M, Sarasin A, Bootsma D, Egly J-M, Hoeijmakers J H J. Cold Spring Harbor Symp Quant Biol. 1994;59:317–329. doi: 10.1101/sqb.1994.059.01.036. [DOI] [PubMed] [Google Scholar]
- 19.Friedberg E C. BioEssays. 1996;18:731–738. doi: 10.1002/bies.950180908. [DOI] [PubMed] [Google Scholar]
- 20.Zawel L, Kumar K P, Reinberg D R. Genes Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- 21.Conaway J W, Conaway R C. Science. 1990;248:1550–1553. doi: 10.1126/science.2193400. [DOI] [PubMed] [Google Scholar]
- 22.Weist D K, Wang D, Hawley D K. J Biol Chem. 1992;267:7733–7744. [PubMed] [Google Scholar]
- 23.Kadesch T R, Chamberlin M J. J Biol Chem. 1982;257:5286–5295. [PubMed] [Google Scholar]
- 24.Donahue B A, Yin S, Taylor J-S, Hanawalt P C. Proc Natl Acad Sci USA. 1994;91:8502–8506. doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sancar A. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 26.Hanawalt P C. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 27.van Gool A J, Verhage R, Swagemakers S M A, van de Putte P, Brouwer J, Troelstra K, Bootsma K, Hoeijmakers J H J. EMBO J. 1994;12:5361–5369. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aso T, Conaway J W, Conaway R C. FASEB J. 1995;9:1419–1428. doi: 10.1096/fasebj.9.14.7589983. [DOI] [PubMed] [Google Scholar]
- 29.Reines D, Conaway J W, Conaway R C. Trends Biochem Sci. 1996;21:351–355. [PMC free article] [PubMed] [Google Scholar]
- 30.Drapkin R, Reardon J R, Ansari A, Huang J C, Zawel L, Ahn K, Sancar A, Reinberg D. Nature (London) 1994;368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- 31.Balajee A S, May A, Dianov G L, Friedberg E C, Bohr V A. Proc Natl Acad Sci USA. 1997;94:4306–4311. doi: 10.1073/pnas.94.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verhage R A. Ph.D. thesis. Leiden, The Netherlands: Erasmus University; 1996. [Google Scholar]
- 33.Verhage R A, van Gool A J, de Groot N, Hoeijmakers J H J, van de Putte P, Brouwer J. Mol Cell Biol. 1996;16:496–502. doi: 10.1128/mcb.16.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper P K, Nouspikel T, Clarkson S G, Leadon S A. Science. 1997;275:990–993. doi: 10.1126/science.275.5302.990. [DOI] [PubMed] [Google Scholar]
- 35.Iyer N, Reagan M S, Wu K J, Canagarajah B, Friedberg E C. Biochemistry. 1996;35:2157–2167. doi: 10.1021/bi9524124. [DOI] [PubMed] [Google Scholar]