

Abstract
Estrogens have been implicated in the regulation of prolactin gene expression in man, although previous studies have not defined the molecular mechanism whereby estradiol activates the human prolactin gene promoter (hPrl). We found that estradiol induced a reproducible 1.8-fold activation of the hPrl gene promoter, using pituitary GH3 cells stably transfected with a 5000-bp hPrl promoter fragment linked to luciferase reporter gene. This activation was blocked by treatment with estrogen receptor (ER) antagonists 4-hydroxytamoxifen and ICI-182,780. Promoter deletion and mutagenesis experiments identified a functional estrogen response element (ERE) sequence 1189 bp upstream of the transcription start site that was responsible for estrogen-mediated promoter activation. This site differed from the consensus ERE sequence by two base pairs, one in each half-site. This ERE was identified to be functional through binding ERα in EMSAs. Chromatin immunoprecipitation assays confirmed ERα binding to this sequence in vivo in the absence of ligand, with increased recruitment when cells were cultured in the presence of estradiol. When cells were treated with both estradiol and TNFα, we observed synergistic activation of the hPrl promoter, which was mediated by the –1189-bp ERE. Mutagenesis of this ERE abolished the promoter-activating effect not only of estradiol but also of TNFα. These data suggest a novel, promoter-specific signaling interaction between estrogen and TNFα signaling, which is likely to be important for prolactin regulation in vivo.
The human prolactin gene (hPrl) is located on the short arm of chromosome 6 (1) and consists of five coding exons within a region of 10 kb. It is expressed in man in lactotroph cells in the anterior pituitary but also in the endometrium and myometrium of the uterus and in T lymphocytes among a number of extrapituitary sites (2-4). In pituitary cells, prolactin gene expression is regulated in response to multiple hormonal signals by an extensive 5′-flanking region extending 5000 bp upstream from the transcriptional start site, which contains multiple binding sites for Pit-1 (5), whereas in extrapituitary tissues, an alternate upstream promoter facilitates expression of an upstream noncoding exon, exon 1a (2, 6).
Pituitary prolactin expression in vivo is regulated by dopamine and other hypothalamic factors such as TRH and by an increasing number of intrapituitary factors (reviewed in Ref. 7). In addition to a series of growth factors, such as fibroblast growth factor-2 (FGF2) and epidermal growth factor, cytokines such as TGFβ and TNFα [acting through nuclear factor-κB (NFκB) activation] are important regulators of lactotroph function (8, 9).
Estrogen has long been regarded as a major physiological activator of prolactin synthesis and lactotroph proliferation (7, 10) and has been implicated in the pathophysiology of hyperprolactinemia and prolactinomas (11). 17β-Estradiol (E2) stimulates lactotroph proliferation and activates prolactin gene expression (12, 13), and rats treated with high doses of estrogen develop pituitary lactotroph hyperplasia (7, 14). E2 exerts its genomic actions through binding estrogen receptors (ERα and ERβ), which are ligand-activated transcription factors residing in the cytosol and translocating to the nucleus upon ligand binding. As with many other steroid hormone receptors, ERs can either directly bind to their consensus target DNA sequence or interact with nuclear coactivators or corepressors, thereby either enhancing or repressing gene expression. Studies in ER knockout mice showed that loss of the ERβ had no effect on Prl mRNA levels in the anterior pituitary, whereas ERα knockout led to loss of Prl expression (15, 16).
In this study, we show that E2 acts through ERα to activate the hPrl promoter through a degenerate estrogen response element (ERE) sequence found at −1189 bp relative to the transcription start site. Mutation of this site abolished estrogen activation of the promoter. This site was able to bind ERα in EMSAs, and chromatin immunoprecipitation (ChIP) assays demonstrated recruitment of ERα, which increased upon E2 treatment of pituitary cells. The transcriptional effects of E2 were significantly amplified by simultaneous treatment with a range of other hormonal stimuli, with a striking synergistic activation when E2 was combined with TNFα. This interaction between estrogen and TNFα signaling was mediated through the −1189-bp ERE and suggests a novel mechanism in the transcriptional regulation of the Prl gene.
Materials and Methods
Plasmids
The hPrl5000luc plasmid used in transient transfections and stably integrated in the GH3/hPrl-luc cell line has been previously described (17). Site mutations were induced in the hPrl5000luc plasmid using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, Cambridge, UK) following the manufacturer's guidelines. Oligonucleotides used for mutation of the −1189-bp ERE site mutation were mutERE sense (S), 5′-GATGAATTTTCCTAACCTAGGCCTCAGAGTGGCTCAG-3′, and mutERE antisense (AS), 5′-CTGAGCCACTCTGAGGCCTAGGTTAGGAAAATTCATC-3′ (underlining represents mutated bases). For in vitro expression of recombinant ERα, the ERα coding sequence was inserted into the pTNT vector (Promega, Southampton, UK) under the control of the T7 promoter.
Cell culture and luciferase assays
Cells were maintained in phenol red-free DMEM with pyruvate/glutamax (Invitrogen, Paisley, UK) and the addition of 10% fetal calf serum (Harlan Sera-Lab, Bicester, UK). With the exception of TNFα, FGF2 (Merck Biosciences) and ICI-182,780 (Tocris), all reagents were obtained from Sigma Chemical Co. (Poole, UK). For transient transfections, 1.2 × 106 GH3 cells were grown in 10-cm dishes for 24 h and then transfected using FuGene 6 (Roche Molecular Biochemicals, West Sussex, UK) according to the protocol of the manufacturer. Initially, 10% of transfected DNA comprised a pcmvβgal reporter used to control for transfection efficiency. However, because the entire population of cells was transfected with a single transfection master mix and subsequently split into individual wells after transfection, we consistently noticed no inter-well variation of βgal expression, indicating equivalent transfection efficiency between replicates. For cotransfection studies, the total amount of DNA used was held constant by addition of empty expression vector. After 24 h, cells were trypsinized and seeded onto 24-well plates. For luciferase assays, 1 × 105 cells per well were seeded in 24-well plates, serum starved for 24 h, and then stimulated as indicated. Cells were washed twice with ice-cold PBS, and lysates (25 mm Tris/PO4 , 10 mm MgCl2, 5 mm EDTA, 15% glycerol, 0.1% Triton X-100, and 0.1 mg/ml BSA) were prepared. The luciferase activity was measured using a Berthold Mithras 960 luminometer (Berthold Technologies, Redbourn, UK). Four to eight replicates were analyzed for each treatment group, and experiments were performed at least three times. Results are shown as mean ± sd fold induction of at least three independent experiments. For statistical analysis, P values were calculated using Student's t test or two-way ANOVA where appropriate.
Primary cultures
Fischer 344 rats were decapitated and anterior pituitaries extracted. The pituitary was then dissected into small fragments and treated with 20 μg/ml DNase I (Sigma) and 50 μg/ml collagenase (Sigma). Cells were resuspended phenol red-free DMEM with pyruvate/glutamax (Invitrogen) and 10% fetal calf serum, counted, and seeded in 24-well plates at a concentration of 1 × 105cells per well. For experiments, the fetal calf serum-containing medium was replaced by serum-free phenol red-free DMEM supplemented with 0.25% BSA after 24 h. After an additional 24-h incubation, cells were stimulated accordingly.
Quantitative PCR (qPCR)
For qPCR analysis, GH3 cells were grown in 25-cm2 flasks (5 × 106 cells), serum starved for 24 h, and then stimulated with 10 nm E2 and/or 10 ng/ml TNFα for the indicated times. Cells were harvested by trypsinization and washed twice in ice-cold PBS. Total RNA was isolated using the QIAGEN (Hilden, Germany) RNeasy kit, and cDNA was generated with the Omniscript RT system (QIAGEN). Quantitative real-time PCR using the Stratagene Mx3000 P thermocycler and the SYBRgreen Jump Start Taq Ready Mix (Sigma) was performed as follows: 10 min at 95 C, followed by 40 cycles of 15 sec at 95 C, 1 min at 56 C, and 30 sec at 72 C. The following primers were used: cyclophilin forward 5′-TTTTCGCCGCTTGCTGCAGAC-3′ and reverse 5′-CACCCTGGCACATGAATCCTGGA-3′; rat prolactin forward 5′-AGCCAAGTGTCAGCCCGGAAAG-3′ and reverse 5′-TGGCCTTGGCAATAAACTCACGA-3′. This resulted in amplicons of 217 and 227 bp, respectively. The dissociation curves of used primer pairs showed a single peak, and samples after PCR had a single DNA band of the expected size in an agarose gel. Cyclophilin was used as a housekeeping gene for normalization. Data are shown as average ± sd of three independent experiments.
EMSA
Recombinant ERα was produced by the TNT in vitro transcription/translation assay (Promega) according to the manufacturer's instructions using a pTNT-ERα plasmid. Electrophoretic assays were performed in a 20-μl reaction containing binding reaction buffer [20 mm Tris (pH 7.5), 1 mm dithiothreitol, 1 mm EDTA, 100 mm KCl, 2 mm MgCl2, 50 μg/ml BSA, and 10% glycerol], 1 μl recombinant ERα, 1 μg polydeoxyinosinic-deoxycytidylic acid, and 20,000 cpm probes labeled with [γ-32P]dATP (Amersham, Piscataway, NJ) using polynucleotide kinase (Roche). For supershift studies and for competition studies, 1 μl ERα HC20-X rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or an excess unlabeled DNA was added to the reaction. Protein-DNA complexes were resolved on a 5% native polyacrylamide gel using 0.5× Tris-borate-EDTA buffer. Gels were then dried and subjected to autoradiography.
ChIP
A total of 2 × 106 GH3/hPrl-luc cells were serum starved for 24 h and treated with 10 nm E2 or 0.1% dimethylsulfoxide (DMSO). Chromatin was cross-linked using 1% formaldehyde for 10 min at 37 C, and the cells were collected after two washings with PBS and lysed in 200 μl ChIP lysis buffer [1% SDS, 10 mm EDTA, 50 mm Tris-HCl (pH 8.1), and protease inhibitor cocktail (Calbiochem, La Jolla, CA)] on ice for 15 min. All subsequent buffers contained the protease inhibitor cocktail. The cell suspension was then sonicated for five 30-sec cycles in a Kerry Pulsatron sonicating bath, with 30 sec on ice between cycles. After centrifugation, 20 μl of the supernatants was retained and used as the input control, and the remainder was diluted to 2 ml in IP buffer [0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl (pH 8.1), 167 mm NaCl]. This diluted fraction was subjected to IP with 4 μg of either anti-Era antibody (HC20; Santa Cruz), anti-Pit-1 (X-7; Santa Cruz), or nonspecific IgG (Santa Cruz) overnight after 2 h preclearing at 4 C with 20 μl preimmune IgG (Sigma), 0.05% BSA, 5 μg sheared salmon sperm DNA, and 5 μl protein A-Dynabeads (Invitrogen). Complexes were recovered by 2 h incubation at 4 C with 2 μg sheared salmon sperm DNA and 2 μl protein A-Dynabeads. Precipitates were serially washed for 15 min with 500 μl buffer I [0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 150 mm NaCl), buffer II [0.1% SDS 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 500 mm NaCl), buffer III [0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-HCl (pH 8.1)] and then twice with Tris-EDTA buffer [1 mm EDTA, 10 mm Tris-HCl (pH 8.0)]. Precipitated chromatin complexes were removed from the beads through a 30-min incubation with 50 μl 1% SDS, 0.1 m NaHCO3, with vortexing each 5 min. This step was repeated twice. Cross-linking was reversed by an overnight incubation at 65 C. DNA was purified with QIAquick PCR purification kit (QIAGEN), as indicated by the manufacturer, and used in a PCR with the following primers: prolactin ERE/Pit-1 forward 5′-AGGCTGCTTTAGATGCATGG-3′ and reverse 5′-TTCTCTGACCCTGAGCCACT-3′; rat GAPDH forward 5′-GAAATGGGCTTAGGGGTGAT-3′ and reverse 5′-TTAAGGATGGCCTTGGACTG-3′.
Results
Estradiol activates the hPrl promoter
To assess whether estrogen exerts an effect on the 5000-bp hPrl promoter, GH3/hPrl-luc cells were treated with increasing doses of E2 and assayed for luciferase activity after 24 h (Fig. 1A). At concentrations of 10−12 m (1 pm) and above, E2 showed a significant induction of reporter gene activity to a maximal induction of 1.8-fold over untreated cells, with an EC50 of 0.02 nm, suggesting that the 5000-bp hPrl promoter is regulated by E2. Stimulation of GH3/hPrl-luc cells with 10 nm E2 over time demonstrated a progressive increase with time, reaching a plateau by 24 h with no further induction observed up to 72 h (Fig. 1B and data not shown). Using qPCR, we confirmed approximately 4-fold activation by E2 of the endogenous rat Prl (rPrl) gene in clonal GH3/hPrl-luc cells (Fig. 1C, upper panel) and in primary cultures of Fischer-344 rat pituitary glands (Fig. 1C, lower panel).
Fig. 1.
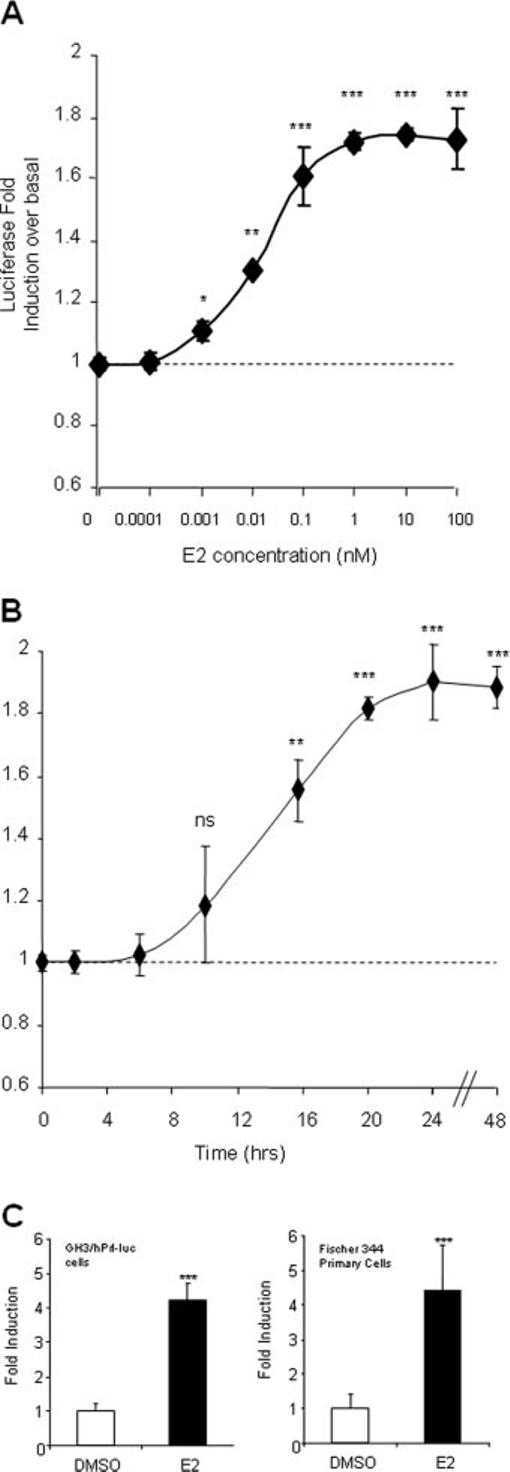
E2 activates the hPrl promoter in GH3/hPRL-LUC cells. Serum-starved GH3/hPrl-luc cells were stimulated with E2, lysed at relevant time points, and assayed for luciferase activity. A, Increasing doses of E2 were used ranging from 0–100 nm for 24 h; B, 10 nm E2 was used and cells lysed at various time points for assay. The dotted line indicates baseline determined from 0.1% DMSO control. C, To verify these cells as a model system for E2-induced Prl gene expression, RNA was extracted, reverse transcribed, and subjected to qPCR for rPrl expression, using rat cyclophilin as an internal control (upper panel). The qPCR was also performed on cDNA from Fischer rat pituitary primary cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001, Student's t test. The data are the mean ± sd of four independent experiments, each with four replicates.
ER-specific antagonists inhibit estradiol induction of the promoter
The selective ER modulator 4-hydroxytamoxifen (4OHT) and the pure estrogen antagonist ICI-182,780 were used to investigate the ER specificity of the estrogen regulation of the hPrl promoter. The antagonists were used at the optimal concentrations, determined using ERE-luc transfected GH3 cells (data not shown), on GH3/hPrl-luc cells treated with 1 nm E2. Treatment with these antagonists abolished reporter gene induction from the hPrl promoter at concentrations of 1000 and 10 nm for 4OHT and ICI-182,780, respectively (Fig. 2A), indicating ER specificity in promoter activation. Dose-response data for the two antagonists showed that ICI-182,780 but not 4OHT significantly reduced luciferase activity to below basal expression levels (Fig. 2B), which may be due to proteasomal mediated degradation of the ER by the ICI antagonist (18). These data suggest a possible role of the ER itself in hPrl transcription, irrespective of ligand.
Fig. 2.
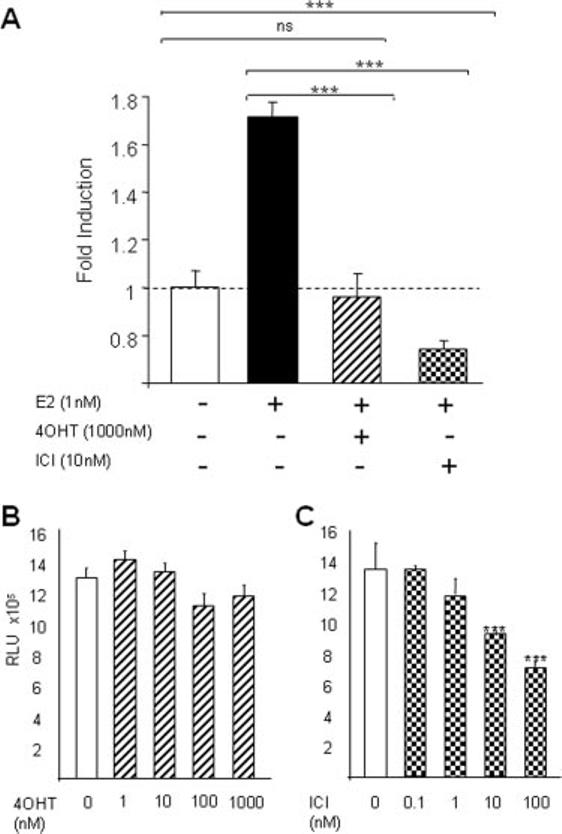
Effects of antagonists on the hPrl promoter in GH3/hPRL-LUC cells. A, Serum-starved GH3/hPrl-luc cells were treated with the indicated stimuli at concentrations of 1 nm E2, 1000 nm 4OHT, and 10 nm ICI-182,780 (ICI) for 24 h. After treatment, cells were lysed and assayed for luciferase activity. The dotted line indicates baseline determined from 0.1% DMSO control. B and C, Starved GH3/hPrl-luc cells were treated with either vehicle (0.1% DMSO) or the indicated concentrations of 4OHT (A) or ICI (B) for 24 h, lysed, and assayed for luciferase activity. ns, Not significant; ***, P < 0.001, Student's t test. The data are the mean ± sd of three independent experiments, each with four replicates.
The hPrl promoter has an ERE at −1189 bp
Previous studies on the rPrl promoter have identified an ERE sequence at −1575 bp (19). This is found adjacent to a Pit-1 binding site within the distal enhancer sequences of the promoter. The corresponding sequence in the hPrl promoter has previously been reported to be functionally different, with no evidence of synergistic cooperation between ER and Pit-1, in contrast to the rat gene (20). The corresponding sequence in the hPrl promoter differs from a consensus ERE sequence by two base pairs, one for each half-site. These two altered bases also diverge from the consensus ERE in the rPrl promoter, although the substitutions are different. In addition, two of the three less conserved intermediate bases between the two half-sites are also altered between the rat and human sequences (Fig. 3A), which has led some authors to suggest that this degeneracy leads to loss of ERE function in the hPrl promoter (20). Several truncated hPrl promoter-luciferase constructs ranging from −4.8 to −0.8 kb were constructed and assessed for E2 responsiveness. All constructs were responsive to E2 except for the shortest (−0.8 kb), indicating estrogen-responsive sequences to be located between −1779 and −828 bp, which includes the putative ERE sequence (Fig. 3B).
Fig. 3.
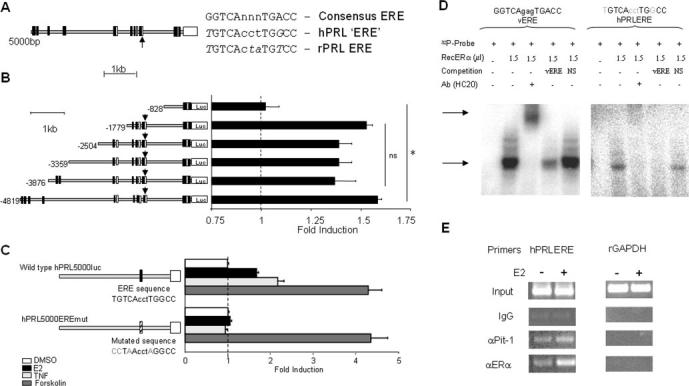
Identification of an ERE in the hPrl promoter. A, Potential EREs within the hPrl promoter are shown by white rectangles, as identified by AliBaba 2.1 (44). Pit-1 binding sites are shown by black rectangles. The arrow indicates the ERE with the closest match to the consensus ERE and corresponds to a previously described ERE within the rPrl promoter (19). Altered bases from consensus are indicated in italics. B, Deletion constructs of the hPrl5000luc plasmid were transfected into GH3 cells. Transfected cells were serum starved for 24 h and treated with 1 nm E2 for 24 h, lysed, and assayed for luciferase activity. The dotted line indicates baseline determined from 0.1% DMSO control. ns, Not significant; *, P < 0.001. C, The −1189-bp ERE site was mutated to abolish any similarity to a consensus sequence. The mutated and wild-type promoter luciferase constructs were transfected into GH3 cells, serum starved for 24 h, treated with 1 nm E2 for 24 h, lysed, and assayed for luciferase activity. D, Binding of ER to a consensus vitellogenin A2 ERE (vERE; left panel) and the −1189-bp ERE of the hPrl promoter (right panel) by EMSA. Specificity was confirmed through antibody supershift (using anti-ERα HC20; Santa Cruz) and competition through excess of unlabeled consensus sequence and nonspecific (NS) DNA sequence. Arrows indicate retarded bands and supershift. E, Serum-starved cells were treated for 24 h with either vehicle (0.1% DMSO) or E2 (10 nm). Cells were fixed in 1% formaldehyde and subjected to ChIP with either a nonspecific IgG antibody, Pit-1-specific antibody X-7, or ERα-specific antibody HC20. DNA was isolated and amplified using primers flanking the hPrl ERE or a nonspecific rGAPDH promoter sequence. Data shown are representative of three separate experiments.
To confirm whether this putative ERE at −1189 bp was functional, we performed site-directed mutagenesis to disrupt the sequence, and the resulting construct was transiently transfected into pituitary GH3 cells (Fig. 3C). Mutation of this sequence resulted in loss of promoter activation by 10 nm E2, consistent with its constituting the functional ERE in the hPrl gene. In control experiments, we found that forskolin, which activates adenylyl cyclase and mediates promoter activation through proximal sequences, stimulated both the wild-type and mutant promoters similarly. Surprisingly, TNFα stimulation of promoter activity was completely abolished by the ERE mutation (Fig. 3C). Previously in our lab, TNFα has been shown to mediate hPrl promoter stimulation through activation of NFκB (9), but despite mutagenesis of several potential NFκB binding sites in the promoter, a critical TNFα-responsive element was not previously defined. The present results suggest that TNFα action may in fact be mediated through a functional ERE.
EMSAs were used to assess whether the hPrl ERE sequence was able to bind ER (Fig. 3D). When incubated with recombinant ERα, the consensus ERE sequence from the vitellogenin A2 gene gave a strong retarded band that was supershifted using the anti-ERα antibody HC20, competed by an excess of unlabeled homologous DNA sequence, but not competed by a nonspecific oligonucleotide (Fig. 3D, left panel). When the same assay was performed using the hPrl ERE sequence, the same general binding pattern was observed with a clear but weaker retarded DNA-protein complex that was supershifted by the anti-ERα antibody and displayed sequence-specific competition (Fig. 3D, right panel). ChIP assays showed recruitment of Pit-1 to this promoter region and confirmed binding of ERα, the level of which appeared to increase when cells were treated with E2 treatment (Fig. 3E).
Interactions between estradiol and TNFα signaling
Due to the finding that TNFα activation of the hPrl promoter was abolished by mutation of the −1189-bp ERE, we investigated the possibility that there is an interaction between estrogen and TNFα signaling. When cells were cotreated with E2 in combination with TRH, FGF2, or forskolin, there was increased induction of the promoter compared with either stimulus alone, roughly equivalent to the sum of the two individual stimulations (data not shown). On the other hand, when TNFα and E2 were used to treat GH3/hPrl-luc cells, a synergistic induction of reporter gene activity was observed. In time-course experiments, E2 and TNFα cotreatment induced synergistic stimulation within 24 h, with more marked synergy apparent by 48–72 h (Fig. 4, A and B). Cell proliferation assays showed that this increase in luciferase activity was not attributable to increased cell number (data not shown). This synergistic transcriptional induction appeared to be specific to the hPrl promoter, because E2 and TNFα costimulation resulted in either no effect or an antagonistic combinatorial effect, respectively, when pituitary GH3 cells were studied after transfection with either a consensus NFκB-luc reporter construct or a consensus ERE-luc construct (Fig. 4C). The synergistic effect was completely abolished when cells were cotreated with the ER antagonists 4OHT and ICI-182,780 (Fig. 4B). Transcription of the endogenous rPrl gene was analyzed by qPCR to assess whether the synergistic interaction of ER and NFκB signaling was an effect confined to the hPrl promoter in the stably transfected cell line. A small additive effect between the two stimuli was observed at 24 h (Fig. 4D, left panel), but a large synergistic interaction (∼83-fold) was seen after 48 h treatment (Fig. 4D, right panel), indicating that the rPrl promoter was affected in a similar manner to the human gene by the combination of these stimuli.
Fig. 4.
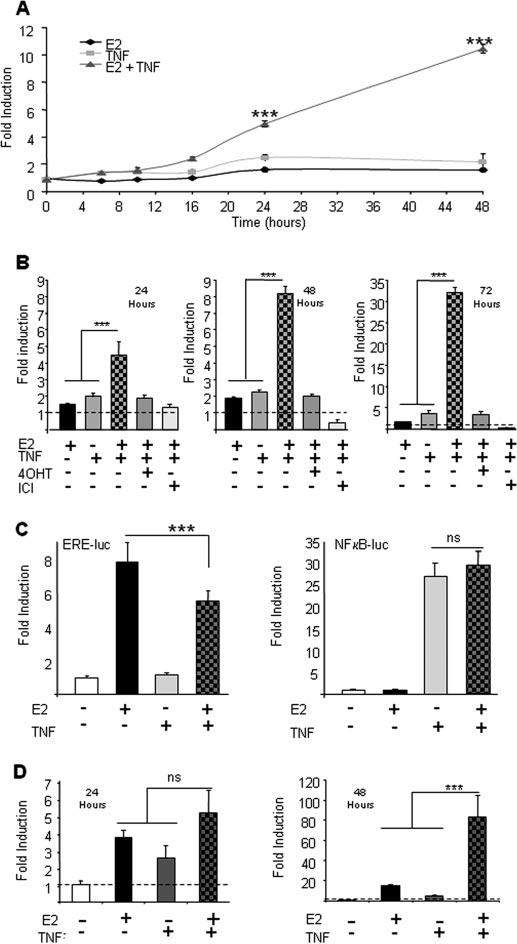
E2 and TNFα synergistically activate the hPrl promoter. A, Serum-starved GH3/hPrl-luc cells were treated with either a 0.1% DMSO control, 1 nm E2, 10 ng/ml TNFα, or E2 and TNFα together for various times, lysed, and assayed for luciferase activity. B, Serum-starved GH3/hPrl-luc cells were treated as indicated [1 nm E2, 10 ng/ml TNFα, 1000 nm 4OHT, and 10 nm ICI-182,780 (ICI)], lysed, and assayed for luciferase activity after 24, 48, or 72 h. The dotted line indicates baseline determined from 0.1% DMSO control (note the change in the y-axis scale). C, ERE-luc and NFκB-luc constructs were transfected into GH3 cells (left and right panels, respectively). Transfected cells were serum starved for 24 h, treated as indicated for 24 h, lysed, and assayed for luciferase activity. ns, Not significant; ***, P < 0.001 examined by two-way ANOVA. The data are the mean ± sd of three independent experiments, each with four replicates. D, Serum-starved GH3/hPrl-luc cells were untreated as indicated for 24 h (left) or 48 h (right). Cells were harvested, and RNA was prepared, reverse transcribed, and subjected to qPCR analysis using primers to examine the expression of rPrl transcript. Data were normalized using expression of the housekeeping gene cyclophilin. ns, Not significant; ***, P < 0.001 examined by two-way ANOVA. The data are the mean ± sd of four independent experiments, each with four replicates.
Discussion
Estrogen is regarded as an important physiological signal in the regulation of the prolactin gene in man, but previous studies have not identified the mechanisms of regulation (20, 21). Here we have identified a degenerate ERE sequence within the human prolactin promoter at −1189 bp relative to the transcription start site, which differs significantly from the rPrl ERE, although it is located within the corresponding enhancer sequence (20, 21). The hPrl ERE differs from a consensus sequence by two bases, one in each half-site, and has lower affinity for binding ERα as shown by in vitro EMSAs. However, ChIP assays clearly showed this ERE to recruit ERα in vivo both in the presence and absence of E2, with possible increase in recruitment after stimulation. This particular ERE sequence also appears to be essential both for the TNFα-induced, NFκB-mediated activation of the hPrl promoter and for the marked synergistic transcriptional effects of estrogen and TNFα that we have identified. Thus, this ERE sequence in the hPrl gene appears to be a target for two converging signaling pathways and may be important for the physiological interpretation of multiple endocrine and stress signals in vivo.
We confirmed that a putative hPrl ERE sequence is indeed a functional estrogen-responsive sequence and suggest that the relatively small effect of E2 alone and the unusual marked synergistic effects observed between E2 and TNFα may be related to the structure of the sequence. The ERE described in the rPrl promoter (19) and the ERE described in this paper both differ from the consensus ERE at the same two bases, but one of these is different in the human sequence, and the intermediate three bases are also different (Fig. 3). Two or more base pair changes from the consensus ERE reduce ERα binding affinity (22) but can lead to altered function through different cofactor recruitment. For example, a coactivator of ERα on the pS2 promoter is steroid receptor coactivator (SRC)-3, involving an ERE with two base changes in the 3′ half-site, whereas the progesterone receptor promoter contains an ERE with four base pair changes in the 3′ half-site and recruits SRC1 and cAMP response element binding protein-binding protein (23). This suggests that the hPrl ERE, with a unique nucleotide sequence, may allow altered function in the actions of ERα for this target gene. The ER may exert some effects on transcription even in the relatively unliganded state. ChIP studies (24, 25) indicate that ERα binds to DNA response elements when unliganded, and it has recently been reported that ERα is associated with the rPrl distal enhancer in the absence of ligand (26). The pure ER antagonist ICI-182,780 caused a reduction in basal hPrl promoter activity (Fig. 2B) and is known to lead to ERα degradation (27), which implies a role for unliganded ERα in the background level of activation of the hPrl promoter even in the absence of specific stimulation by E2.
The pituitary-specific transcription factor Pit-1 is likely to have an important role in estrogen action on the Prl gene (28, 29). We found that Pit-1 bound to the hPrl gene region containing the −1189-bp ERE in both the presence and absence of E2 (Fig. 3E), which concurs with previous studies on the rPrl promoter (30) and may indicate that Pit-1 and ER are involved in the formation of a multiprotein complex at this site. Liu et al. (30) did not detect changes in Pit-1 association/dissociation with the rPrl promoter after treatment with various hormones or agents. The authors proposed that Pit-1 functions in part as a promoter-bound docking site for co-regulatory complexes recruited by diverse hormone signals. Furthermore, it has been reported that Pit-1 is essential for the nucleosomal positioning within the rPrl promoter and contributed to the mechanisms that establish the cell-specific chromatin structure of the Prl gene (31). Previous work has suggested that ERα and Pit-1 exert synergistic transcriptional effects on the rat but not the hPrl promoter (20). However, that study was performed using chimeric promoter fragments expressed in the human uterine cell line SKUT-1B-20. This cell line does not endogenously express Pit-1 or ERα, and therefore the expression of these proteins was manipulated by transient transfection with expression vectors. The level of transcription factor expression is normally highly regulated and the available concentration of transcription factor is therefore likely to influence gene regulation (32, 33). The cell lines we used are derived from somatolactotroph cells of the anterior pituitary and are likely to replicate endogenous levels of protein expression more authentically than the SKUT-1B-20 cell line. By using constructs containing natural intact promoter sequences, our cell model may therefore represent more accurately the regulation of the hPrl gene than previous studies.
TNFα has been shown to activate hPrl gene transcription via NFκB signaling (9). The precise DNA sequences involved in this activation were not identified despite mutagenesis of several putative NFκB binding sites. In the present study, mutagenesis of the hPrl ERE completely blocked not only the activating effect of estrogen but also that of TNFα, indicating that NFκB activation of the promoter is mediated through the ERE and suggesting an interaction between the two signaling pathways. Steroid hormone receptors, including ERα, have long been described to interact with NFκB, usually in an antagonistic fashion, with the mechanism of antagonism depending on both the cell type (34) and the promoter (35). However, in our system, cotreatment of TNFα and E2 in GH3/hPrl-luc cells led to a synergistic activation of reporter gene activity. This may be a promoter-specific effect whereby ER and NFκB can be members of the same regulatory complex due to the unique ERE sequence found at −1189 bp. The qPCR analysis confirmed a similar synergistic interaction between E2 and TNFα signaling in activation of endogenous rPrl gene expression. Such a synergistic interaction between these signals was not, however, seen using either consensus ERE or NFκB response elements linked to luciferase reporter genes in GH3 cells, indicating that the effect was Prl promoter specific (Fig. 4C). In fact, an antagonistic effect was observed on the transfected ERE-luc construct, reminiscent of the widely reported mutual antagonism of NFκB and nuclear hormone receptors observed in other systems (35, 36).
The synergistic activation of the hPrl promoter by E2 and TNFα could be blocked by tamoxifen, with a reduction of activity to the level of stimulation induced by TNFα alone, implying that the interactive effect of E2 and TNFα requires the AF-2 domain of ERα and potentially requires recruitment of coactivators (37, 38). ICI-182,780 is regarded as a pure ER antagonist and can lead to proteasomal degradation of the ER (27). Reid et al. (39) reported constitutive, low-level recruitment of ERα to the pS2 promoter that increased upon treatment with E2. However, on treatment of cells with ICI-182,780, ER recruitment was lost within 3 h (irrespective of the presence or absence of E2), with evidence for its proteasomal degradation, and such receptor turnover may be an integral feature of estrogen signaling (39). In the present work in pituitary cells, the ICI antagonist not only blocked the synergistic effect of E2 and TNFα but also reduced promoter activity to well below basal levels, suggesting that ER itself is essential to maintain Prl basal expression. In view of these findings, we propose that the TNFα stimulatory effects on the hPrl promoter require the presence of ERα, irrespective of the presence of estrogen, and that addition of E2 ligand permits further increases in transcription. Given the promoter-specific nature of this cross-talk, Pit-1 may be necessary to facilitate such an interaction, although this requires more investigation.
Few studies describe positive interactions between ERα and NFκB. Rubio et al. (40) showed ER and NFκB to have combinatorial effects on genes controlling cell division in ER-positive breast cancer cells, with ChIP assays showing recruitment of both ERα and NFκB to the cyclin D1 promoter. Although direct interaction between the two proteins was not determined, the coactivator SRC3, which has been described as a coactivator in gene activation by both ER (41) and NFκB (42), was found to be interacting with both the NFκB subunit p65 and ERα in co-IP experiments, suggesting that interactions between these two signaling pathways may be indirect and require a complex formation with other transcription factors (40). Wissink et al. (43) showed the serotonin 1A receptor promoter to be synergistically activated by p50/p65 (NFκB) and ERα in COS-1 cells. The effect occurred with unliganded ER and was further increased upon addition of E2. Interestingly, the synergistic interaction observed on the serotonin 1A receptor promoter was mediated through degenerate NFκB binding sites, and not an ERE as in the hPrl promoter.
The role of TNFα within the pituitary is unknown but may mediate paracrine or inflammatory effects (9). E2 is reported to have several different endocrine effects within the pituitary, including regulation of multiple genes and secretion of gene products (7). Here we show the signaling pathways from these two important stimuli to interact, indicating that TNFα signaling may be an important amplifier of estrogen response. It would be of interest to study this in vivo to analyze fully the endocrine role of TNFα in modulating other signaling pathways in the context of the full endogenous locus.
In summary, we have identified an ERE in the hPrl gene and have found a novel synergistic interaction between estrogen and cytokine signaling that facilitates increased expression of hPrl. The ERE is unusual and is necessary also for TNFα activation of the gene and to mediate the synergistic interaction between these two converging signal pathways. It is likely that estrogen, while having a relatively small effect in isolation, is a critical factor that may dramatically amplify prolactin expression in response to other endocrine or paracrine signals in the pituitary.
Acknowledgments
We thank Nicola Halliwell and Michael Wilding for technical assistance and the Wellcome Trust and Medical Research Council for funding this work.
This work was supported by Wellcome Trust Program Grant 067252 and Medical Research Council Ph.D. studentship to A.D.A.
Abbreviations
- ChIP
Chromatin immunoprecipitation
- DMSO
dimethylsulfoxide
- E2
17β-estradiol
- ER
estrogen receptor
- ERE
estrogen response element
- FGF2
fibroblast growth factor-2
- hPrl
human prolactin
- NFκB
nuclear factor-κB
- 4OHT
4-hydroxytamoxifen
- qPCR
quantitative PCR
- rPrl
rat prolactin
- SRC
steroid receptor coactivator
Footnotes
Disclosure Statement: The authors have nothing to disclose.
References
- 1.Owerbach D, Rutter WJ, Cooke NE, Martial JA, Shows TB. The prolactin gene is located on chromosome 6 in humans. Science. 1981;212:815–816. doi: 10.1126/science.7221563. [DOI] [PubMed] [Google Scholar]
- 2.Gellersen B, Kempf R, Telgmann R, DiMattia GE. Nonpituitary human prolactin gene transcription is independent of Pit-1 and differentially controlled in lymphocytes and in endometrial stroma. Mol Endocrinol. 1994;8:356–373. doi: 10.1210/mend.8.3.8015553. [DOI] [PubMed] [Google Scholar]
- 3.Pellegrini I, Lebrun JJ, Ali S, Kelly PA. Expression of prolactin and its receptor in human lymphoid cells. Mol Endocrinol. 1992;6:1023–1031. doi: 10.1210/mend.6.7.1508218. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr Rev. 1996;17:639–669. doi: 10.1210/edrv-17-6-639. [DOI] [PubMed] [Google Scholar]
- 5.Van De Weerdt C, Peers B, Belayew A, Martial JA, Muller M. Far upstream sequences regulate the human prolactin promoter transcription. Neuroendocrinology. 2000;71:124–137. doi: 10.1159/000054528. [DOI] [PubMed] [Google Scholar]
- 6.Berwaer M, Martial JA, Davis JR. Characterization of an up-stream promoter directing extrapituitary expression of the human prolactin gene. Mol Endocrinol. 1994;8:635–642. doi: 10.1210/mend.8.5.8058071. [DOI] [PubMed] [Google Scholar]
- 7.Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80:1523–1631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- 8.Hentges S, Sarkar DK. Transforming growth factor-β regulation of estradiol-induced prolactinomas. Front Neuroendocrinol. 2001;22:340–363. doi: 10.1006/frne.2001.0220. [DOI] [PubMed] [Google Scholar]
- 9.Friedrichsen S, Harper CV, Semprini S, Wilding M, Adamson AD, Spiller DG, Nelson G, Mullins JJ, White MRH, Davis JRE. Tumor necrosis factor-α activates the human prolactin gene promoter via nuclear factor-κB signaling. Endocrinology. 2006;147:773–781. doi: 10.1210/en.2005-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kansra S, Yamagata S, Sneade L, Foster L, Ben-Jonathan N. Differential effects of estrogen receptor antagonists on pituitary lactotroph proliferation and prolactin release. Mol Cell Endocrinol. 2005;239:27–36. doi: 10.1016/j.mce.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Heaney AP, Horwitz GA, Wang Z, Singson R, Melmed S. Early involvement of estrogen-induced pituitary tumor transforming gene and fibro-blast growth factor expression in prolactinoma pathogenesis. Nat Med. 1999;5:1317–1321. doi: 10.1038/15275. [DOI] [PubMed] [Google Scholar]
- 12.Shupnik MA, Baxter LA, French LR, Gorski J. In vivo effects of estrogen on ovine pituitaries: prolactin and growth hormone biosynthesis and messenger ribonucleic acid translation. Endocrinology. 1979;104:729–735. doi: 10.1210/endo-104-3-729. [DOI] [PubMed] [Google Scholar]
- 13.Amara JF, Van Itallie C, Dannies PS. Regulation of prolactin production and cell growth by estradiol: difference in sensitivity to estradiol occurs at level of messenger ribonucleic acid accumulation. Endocrinology. 1987;120:264–271. doi: 10.1210/endo-120-1-264. [DOI] [PubMed] [Google Scholar]
- 14.Lloyd RV, Cano M, Landefeld TD. The effects of estrogens on tumor growth and on prolactin and growth hormone mRNA expression in rat pituitary tissues. Am J Pathol. 1988;133:397–406. [PMC free article] [PubMed] [Google Scholar]
- 15.Scully KM, Gleiberman AS, Lindzey J, Lubahn DB, Korach KS, Rosenfeld MG. Role of Estrogen receptor-α in the anterior pituitary gland. Mol Endocrinol. 1997;11:674–681. doi: 10.1210/mend.11.6.0019. [DOI] [PubMed] [Google Scholar]
- 16.Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) null mice reveals hypergonadism and endocrine sex reversal in females lacking ERα but not ERβ. Mol Endocrinol. 2003;17:1039–1053. doi: 10.1210/me.2002-0398. [DOI] [PubMed] [Google Scholar]
- 17.Takasuka N, White MRH, Wood CD, Robertson WR, Davis JRE. Dynamic changes in prolactin promoter activation in individual living lactotrophic cells. Endocrinology. 1998;139:1361–1368. doi: 10.1210/endo.139.3.5826. [DOI] [PubMed] [Google Scholar]
- 18.Preisler-Mashek MT, Solodin N, Stark BL, Tyriver MK, Alarid ET. Ligand-specific regulation of proteasome-mediated proteolysis of estrogen receptor-α. Am J Physiol Endocrinol Metab. 2002;282:E891–E898. doi: 10.1152/ajpendo.00353.2001. [DOI] [PubMed] [Google Scholar]
- 19.Murdoch FE, Byrne LM, Ariazi EA, Furlow JD, Meier DA, Gorski J. Estrogen receptor binding to DNA: affinity for nonpalindromic elements from the rat prolactin gene. Biochemistry. 1995;34:9144–9150. doi: 10.1021/bi00028a025. [DOI] [PubMed] [Google Scholar]
- 20.Gellersen B, Kempf R, Telgmann R, DiMattia GE. Pituitary-type transcription of the human prolactin gene in the absence of Pit-1. Mol Endocrinol. 1995;9:887–901. doi: 10.1210/mend.9.7.7476971. [DOI] [PubMed] [Google Scholar]
- 21.Berwaer M, Monget P, Peers B, Mathy-Hartert M, Bellefroid E, Davis JR, Belayew A, Martial JA. Multihormonal regulation of the human prolactin gene expression from 5000 bp of its upstream sequence. Mol Cell Endocrinol. 1991;80:53–64. doi: 10.1016/0303-7207(91)90142-f. [DOI] [PubMed] [Google Scholar]
- 22.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klinge CM, Jernigan SC, Mattingly KA, Risinger KE, Zhang J. Estrogen response element-dependent regulation of transcriptional activation of estrogen receptors α and β by coactivators and corepressors. J Mol Endocrinol. 2004;33:387–410. doi: 10.1677/jme.1.01541. [DOI] [PubMed] [Google Scholar]
- 24.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 25.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Sharp ZD, Mancini MG, Hinojos CA, Dai F, Berno V, Szafran AT, Smith KP, Lele TT, Ingber DE, Mancini MA. Estrogen-receptor-α exchange and chromatin dynamics are ligand- and domain-dependent. J Cell Sci. 2006;119:4101–4116. doi: 10.1242/jcs.03161. [DOI] [PubMed] [Google Scholar]
- 27.Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, “pure” antiestrogen. Cancer. 2000;89:817–825. doi: 10.1002/1097-0142(20000815)89:4<817::aid-cncr14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Day RN, Koike S, Sakai M, Muramatsu M, Maurer RA. Both Pit-1 and the estrogen receptor are required for estrogen responsiveness of the rat prolactin gene. Mol Endocrinol. 1990;4:1964–1971. doi: 10.1210/mend-4-12-1964. [DOI] [PubMed] [Google Scholar]
- 29.Nowakowski BE, Maurer RA. Multiple Pit-1-binding sites facilitate estrogen responsiveness of the prolactin gene. Mol Endocrinol. 1994;8:1742–1749. doi: 10.1210/mend.8.12.7708061. [DOI] [PubMed] [Google Scholar]
- 30.Liu JC, Baker RE, Chow W, Sun CK, Elsholtz HP. Epigenetic mechanisms in the dopamine D2 receptor-dependent inhibition of the prolactin gene. Mol Endocrinol. 2005;19:1904–1917. doi: 10.1210/me.2004-0111. [DOI] [PubMed] [Google Scholar]
- 31.Kievit P, Maurer RA. The pituitary-specific transcription factor, Pit-1, can direct changes in the chromatin structure of the prolactin promoter. Mol Endocrinol. 2005;19:138–147. doi: 10.1210/me.2004-0016. [DOI] [PubMed] [Google Scholar]
- 32.Dasen JS, Rosenfeld MG. Signaling and transcriptional mechanisms in pituitary development. Annu Rev Neurosci. 2001;24:327–355. doi: 10.1146/annurev.neuro.24.1.327. [DOI] [PubMed] [Google Scholar]
- 33.Fowler AM, Solodin NM, Valley CC, Alarid ET. Altered target gene regulation controlled by estrogen receptor-α concentration. Mol Endocrinol. 2006;20:291–301. doi: 10.1210/me.2005-0288. [DOI] [PubMed] [Google Scholar]
- 34.Cerillo G, Rees A, Manchanda N, Reilly C, Brogan I, White A, Needham M. The oestrogen receptor regulates NFκB and AP-1 activity in a cell-specific manner. J Steroid Biochem Mol Biol. 1998;67:79–88. doi: 10.1016/s0960-0760(98)00078-8. [DOI] [PubMed] [Google Scholar]
- 35.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends Endocrinol Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 36.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endo Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 37.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 38.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 39.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 40.Rubio MF, Werbajh S, Cafferata EG, Quaglino A, Colo GP, Nojek IM, Kordon EC, Nahmod VE, Costas MA. TNF-α enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-κB. Oncogene. 2006;25:1367–1377. doi: 10.1038/sj.onc.1209176. [DOI] [PubMed] [Google Scholar]
- 41.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-κB requires multiple coactivators. Mol Cell Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werbajh S, Nojek I, Lanz R, Costas MA. RAC-3 is a NF-κB coactivator. FEBS Lett. 2000;485:195–199. doi: 10.1016/s0014-5793(00)02223-7. [DOI] [PubMed] [Google Scholar]
- 43.Wissink S, van der Burg B, Katzenellenbogen BS, van der Saag PT. Synergistic activation of the serotonin-1A receptor by nuclear factor-κB and estrogen. Mol Endocrinol. 2001;15:543–552. doi: 10.1210/mend.15.4.0629. [DOI] [PubMed] [Google Scholar]
- 44.Grabe N. AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol. 2002;2:S1–S15. [PubMed] [Google Scholar]