

Abstract
We investigated whether enhanced cardiac vagal responsiveness elicited by exercise training is dependent on neuronal nitric oxide synthase (NOS-1), since the NO-cGMP pathway facilitates acetylcholine release. Isolated atria with intact right vagal innervation were taken from male mice (18-22 weeks old) after a period of 10 weeks voluntary wheel-running (+EX, n = 27; peaked 9.8 ± 0.6 km day−1 at 5 weeks), and from mice housed in cages without wheels (-EX, n = 27). Immunostaining of whole atria for NOS-1 identified intrinsic neurones, all of which co-localized with choline acetyltransferase-positive ganglia. Western blot analysis confirmed that NOS-1 protein level was significantly greater in +EX compared to -EX atria (P < 0.05, unpaired t test). Basal heart rates (HR) were slower in +EX than in -EX atria (322 ± 6 versus 360 ± 7 beats min−1; P < 0.05, unpaired t test) However, in +EX atria, HR responses to vagal stimulation (VNS, 3 and 5 Hz) were significantly enhanced compared to -EX atria (3 Hz, +EX: −76 ± 8 beats min−1versus -EX: −62 ± 7 beats min−1; 5 Hz, +EX: −106 ± 4 beats min−1versus -EX: −93 ± 3 beats min−1; P < 0.01, unpaired t test). Inhibition of NOS-1 with vinyl-l-N-5-(1-imino-3-butenyl)-l-ornithine (l-VNIO, 100 μm) or soluble guanylyl cyclase with 1H-[1, 2, 4]oxadiazolo[4, 3-a]quinoxalin-1-one (ODQ, 10 μm) abolished the difference in HR responses to VNS between +EX and -EX atria, and effects of l-VNIO were reversed by excess l-arginine (1 mm; P < 0.01, ANOVA). There were no differences between the HR responses to the bath-applied acetylcholine analogue carbamylcholine chloride in +EX and -EX atria (IC50 concentrations were 5.9 ± 0.4 μm (-EX) and 5.7 ± 0.4 μm (+EX)), suggesting that the changes in vagal responsiveness resulted from presynaptic facilitation of neurotransmission. In conclusion, NOS-1 appears to be a key protein in generating the cardiac vagal gain of function elicited by exercise training.
Aerobic exercise training is associated with an increased cardiac vagal responsiveness (Imai et al. 1994; Yamamoto et al. 2001; Sugawara et al. 2001). However, the mechanism underlying the development of the exercise-trained cardiac vagal phenotype is not known (Buch et al. 2002). Exercise training increases expression of endothelial nitric oxide (NO) synthase (NOS-3) in coronary vasculature (Sessa et al. 1994), and neuronal NOS (NOS-1) in cardiac sympathetic ganglia (Mohan et al. 2000). Functionally, NO augments heart rate (HR) responses to vagal nerve stimulation (VNS) in vitro (e.g. Sears et al. 1998; Herring et al. 2000) and in vivo (e.g. Elvan et al. 1997; Conlon & Kidd, 1999) where the NO-cyclic guanosine monophosphate (NO-cGMP) pathway facilitates cardiac cholinergic neurotransmission in a highly isoform- and site-specific fashion (Herring & Paterson, 2001). NOS-1 co-localizes with choline acetyltransferase (ChAT) in intracardiac parasympathetic ganglia (Calupca et al. 2000; Choate et al. 2001), and NOS-1 null mice have impaired HR responses to VNS, but not to bath-applied ACh (Choate et al. 2001). In contrast, NOS-3 resident in cardiac myocytes appears to have little appreciable role in the peripheral autonomic regulation of cardiac excitability (Vandecasteele et al. 1999; Brunner et al. 2001), suggesting that the main functional isoform for cholinergic modulation of rate is NOS-1 and resides presynaptically at the level of the vagal neurone. Here we show that the functional changes in vagal neurotransmission in the mouse heart caused by exercise training result from upregulation of NOS-1.
Methods
Animals
All experiments were performed in accordance with the Animals (Scientific Procedures) Act 1986 (UK, Home Office license requirements PPL 30/1735, Queen Anne's Gate, London, UK). Adult male C57BL/6J mice (8-12 weeks old, purchased from Charles River Ltd, UK) and homozygote NOS-1 knockout mice (8-12 weeks old, purchased from Jackson Laboratories, ME, USA) were used.
Training protocol
Mice were housed singly in cages with (+EX, n = 27) or without wheels (-EX, n = 27). Voluntary running distances covered by +EX mice over a 24 h period were calculated daily from tripometer counters (Cat Eye Electronics, Japan) between 1800 and 2000 h for a training period of 10 weeks. Mean weekly distances were also calculated from daily distances covered by each mouse, to ensure mice had reached their optimum running performance before the 10 week period had elapsed.
Body weights of +EX and -EX mice were measured weekly over the training period. Ventricular weights were measured post-mortem and ventricle:body weight ratios were calculated as an independent index of training.
Immunohistochemistry
Mice (3 -EX; 3 +EX; 2 NOS-1(-/-)) were terminally anaesthetized (pentobarbitone i.p.), and then fixed by perfusion of the left ventricle with 4 % paraformaldehyde and 0.1 % glutaraldehyde in 0.1 m phosphate buffer solution (PBS, pH 7.1, 20 min). The posterior aspect of the right atrium was dissected free and was treated for immunohistochemistry as has been previously described (Wang & Morris. 1996). After processing, the tissue was incubated in primary antiserum against NOS-1 (raised in sheep against whole recombinant rat NOS-1, 1:400 dilution, a gift from Dr P. C. Emson and Dr I. Charles) and primary antiserum against choline acetyltransferase (ChAT, raised in goat, 1:250 dilution, obtained from Vector Laboratories, UK) for 12 h. Tissue was then washed in 1 % chicken egg albumin PBS solution (30 min) and rhodamine-conjugated anti-goat (1:200) and fluorescein-conjugated anti-sheep (1:200) antisera were used as secondary antisera. Immunoreactivity was viewed using confocal microscopy. Atria from NOS-1(-/-) mice were studied as a control for the anti-NOS-1 antibody.
In vitro protocols
Mice (n = 24 +EX, n = 24 -EX) were killed by cervical dislocation. Double-atrial preparations were dissected free with intact right vagus nerve and left for a 1-2 h equilibration period in mouse physiological saline, as previously described (Choate et al. 2001). Experiments were coded before starting the experiment and data were analysed blind.
Vagal nerve stimulation
Changes in heart rate in response to right vagal stimulation at 3 and 5 Hz (10 V, 1 ms duration, 30 s, in random order) were determined. The average of three heart rate responses was determined for each stimulation frequency.
Bath-applied carbamylcholine chloride
In order to assess changes in the postsynaptic regulation of heart rate, the chronotropic responses to cumulative doses of the ACh analogue carbamylcholine chloride (CCh) (0.01-100 μm), which gave similar HR changes as with VNS, were investigated.
Drugs
Vinyl-l-N-5-(1-imino-3-butenyl)-l-ornithine (l-VNIO) was obtained from Calbiochem Novabiochem (Nottingham, UK). Atropine, carbamylcholine chloride (CCh), 1H-[1, 2, 4]oxadiazolo[4, 3-a]quinoxalin-1-one (ODQ), sodium nitroprusside (SNP) and 8-Br-cGMP were obtained from Sigma. All solutions were made up immediately prior to use with a physiological saline as solvent.
Effects of exercise on heart rate responses
In vitro HR responses to VNS and CCh were compared in +EX (n = 24) and -EX (n = 24) atria.
Effects of NOS-1 inhibition on heart rate responses
VNS and CCh protocols were then commenced following 20 min equilibration with a NOS inhibitor. l-VNIO (100 μm) was used as a selective inhibitor of NOS-1 (l-VNIO: Ki[NOS-1] = 90 nm; Ki[NOS-3] = 12 μm; Babu & Griffith, 1998). Excess substrate for NOS (l-arginine, 1 mm, 20 min equilibration) was used to reverse the effect of NOS inhibition.
Effects of a NO donor, soluble guanylyl cyclase inhibition and a cyclic GMP analogue
A NO donor was then used to bypass the endogenous production of NO to determine whether the downstream signalling pathway was altered by training (VNS and CCh protocols). Low doses of SNP (10 μm) release NO and stimulate guanylyl cyclase, rather than causing nitrosylation or the generation of superoxide radicals which is a property of many other donors (Sarkar et al. 2000).
Following wash-off of SNP, the VNS and CCh protocols were repeated in the presence of the soluble guanylyl cyclase inhibitor ODQ (10 μm, 40 min equilibration) to establish whether the effects of NO were likely to be via the cyclic GMP pathway. The cGMP analogue (8-Br-cGMP, 0.5 mm) was then added to the organ bath to determine the effects on vagal bradycardia.
Western blot analysis
Right atria (+EX, n = 6; -EX, n = 4) were rapidly preserved in liquid nitrogen following the completion of the in vitro protocols and lysed in buffer containing 50 mm Hepes (pH 7.4), 1 mm EDTA, 0.5 % Triton-X-100, and a cocktail of protease inhibitors (Sigma). Protein concentrations were measured using the Bio-Rad DC protein assay kit. Then 17.5 μg of total protein was separated on a 4-20 % gradient gel (Criterion, Bio-Rad) at 150 V for 60 min and resolved proteins transferred to Micron Separations Inc. polyvinylidene fluoride membrane (Genetic Research Instrumentation, UK) using a semi-dry transfer cell following manufacturer's protocols (Bio-Rad). Membranes were blocked for 2 h in 6 % dried milk in PBS-0.1 % Tween-20 (PBST) at room temperature, and then incubated overnight in primary antiserum (1:1000, rabbit pAb anti-NOS-1; 1:1000 rabbit pAb anti-eGFP; and 1:200 rabbit pAb anti-β-actin, Vector Laboratories, UK)-PBST solution at 4 °C. Blots were washed three times for 15 min in PBST, incubated with secondary HRP-conjugated antiserum for 1 h at room temperature, and then washed as before. Immunoreactivity was detected using luminol-based chemiluminescence detection reagents (Western Lightening, NEN Life Sciences, UK) and autoradiography. Images were then digitized for analysis.
Statistics
Data are presented as means ± standard error of the mean. All data were normally distributed. Between-group comparisons were performed by Student's unpaired t test. Multiple comparisons within groups were performed by repeated-measures one-way ANOVA, followed by Student-Neumann-Keul's multiple comparison post hoc tests. Statistical significance was accepted at P < 0.05.
Results
Indices of training
Average daily running distances and body weights of mice at the end of weeks 1-10 are shown in Fig. 1. At the end of the 10th week, body weights were reduced (see Fig. 1B, P < 0.01 unpaired t test) and ventricle/body weight ratios were significantly increased in +EX compared to -EX mice (ventricle/body weight ratios: -EX, 5.00 ± 0.09 mg g−1; +EX, 6.02 ± 0.16 mg g−1; P < 0.001, unpaired t test). In vitro intrinsic heart rates were also significantly reduced in +EX atria compared to -EX atria (-EX: 360 ± 7 beats min−1, n = 24versus +EX: 322 ± 6 beats min−1, n = 24; P < 0.01, unpaired t test), indicating a training bradycardia.
Figure 1. Daily distances covered and body weights of mice over training period.
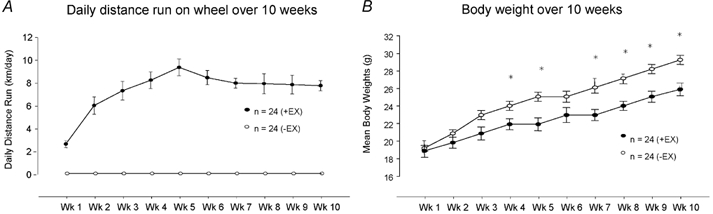
A, average daily distance run by singly housed C57BL/6J mice over a 10-week training period (+EX, •) compared to mice in cages without wheels (-EX, ○). B, average weekly body weight was significantly decreased in +EX compared to -EX mice 4 weeks into the training period (*P < 0.01, unpaired t test). Measurements were taken at the end of each week.
Immunohistochemistry and Western blot analysis of NOS isoform expression
NOS-1-positive neurones co-localized with intracardiac cholinergic ganglia in murine atria. No other cell types or structures stained for NOS-1 in the vicinity of either right or left atrium. Co-localization of NOS-1 is demonstrated in normal atria (+EX, n = 3, Fig. 2A and B; -EX, n = 3, Fig. 2C and D). In -EX atria, 14.4 ± 3.1 % of cholinergic cell bodies co-stained for NOS-1 and the respective figure for +EX atria was 16.5 ± 1.2 %, although this difference failed to reach statistical significance. In NOS-1(-/-) atria, no immunoreactivity against NOS-1 was observed (n = 2; Fig. 2E and F), suggesting that the antibody did not cross-react with other NOS isoforms or splice variants of NOS-1 that may be expressed from exon 6 of the transgene. Western blot analysis of protein extracted from atria revealed a 76 % increase in expression of NOS-1 following training (-EX, n = 4; +EX, n = 6; see Fig. 3A and B, P < 0.05 unpaired t test).
Figure 2. Confocal images showing co-localization of cholinergic ganglia and NOS-1.
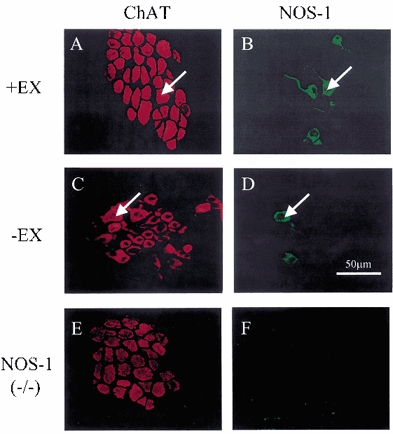
A, cholinergic ganglia in trained (+EX) murine atria visualized by immunoreactivity against choline acetyltransferase (ChAT). B, co-localization of neuronal nitric oxide synthase (NOS-1)-positive neurones in +EX atria. Arrows indicate the same cell in A and B. C, ganglia in control atria (-EX) with co-staining of NOS-1 (D). Arrows indicate the same cell in C and D. E, cholinergic ganglia in the NOS-1(-/-) mouse right atrium. F, NOS-1 co-localization is absent in the NOS(-/-) mouse atrium.
Figure 3. Western blot analysis of NOS-1 expression.
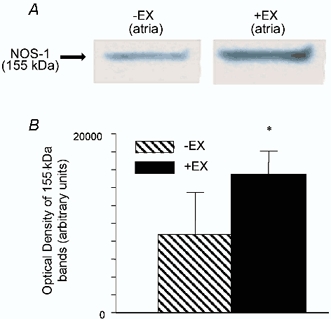
A, Western blot of atrial tissue from trained (+EX) and untrained (-EX) atria with protein bands at 155 kDa (NOS-1). There was a significant increase in NOS-1 expression (*P < 0.05, unpaired t test). β-Actin levels were equal across all bands. B, graph shows the quantitative increase in density of the NOS-1 bands in +EX compared to -EX atria. There was a 76 % increase in protein expression.
Effects of exercise on heart rate responses
The magnitude of HR response to VNS was greater in +EX than -EX atria at both 3 Hz (+EX: −76 ± 8 beats min−1; -EX: −62 ± 7 beats min−1; P < 0.05, unpaired t test) and 5 Hz stimulation frequencies (see Fig. 4). This difference was seen irrespective of whether data were expressed as HR change or RR interval change, which encompasses the fact +EX atria started at a lower intrinsic HR (3 Hz VNS: 0.1654 ± 0.0027 to 0.1974 ± 0.0014 ms in -EX and 0.1770 ± 0.0034 to 0.2330 ± 0.0022 ms in +EX; P < 0.01, unpaired t test; 5 Hz VNS: 0.1663 ± 0.0029 to 0.2239 ± 0.0027 ms in -EX and 0.1828 ± 0.0035 to 0.2802 ± 0.0053 ms in +EX; P < 0.01, unpaired t test). In contrast, HR responses to CCh (0.01-100 μm) were not different in -EX vs. +EX (see Fig. 5). This indicates that the rate response caused by training occurs presynaptically.
Figure 4. In vitro HR changes in response to VNS following NOS-1 inhibition.
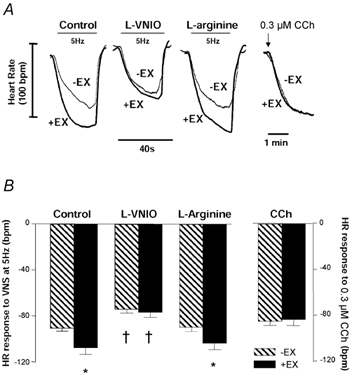
A, raw data traces show heart rate (HR; bpm = beats min−1) changes with vagal nerve stimulation (VNS) at 5 Hz. HR responses were increased in +EX compared to -EX atria. Responses were normalized in +EX with respect to -EX atria following NOS-1 inhibition with l-VNIO (100 μm) - an effect which was reversed by excess l-arginine. HR responses to carbamylcholine chloride (CCh, 0.3 μm) were not different in -EX and +EX. B, graph shows quantitative changes in HR with VNS or CCh. (*P < 0.05, unpaired t test; †P < 0.01, repeated-measures ANOVA).
Figure 5. In vitro HR changes in response to CCh in -EX and +EX atria.
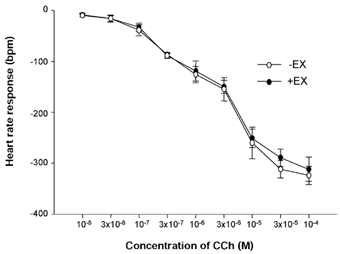
Heart rate (HR) changes in response to doses of carbamylcholine chloride (CCh) were not different in -EX and +EX atria.
Effects of NOS inhibition on heart rate responses
NOS inhibition attenuated HR responses to VNS in +EX and -EX both at 3 Hz (-EX[l-VNIO]: from −62 ± 7 to −47 ± 4 beats min−1; +EX[l-VNIO]: from −76 ± 8 to −48 ± 7 beats min−1; P < 0.01 ANOVA) and at 5 Hz (see Fig. 4). The effects of l-VNIO were reversed by excess l-arginine. Neither l-VNIO nor l-arginine altered basal HR.
Effects of a NO donor, soluble guanylyl cyclase inhibition and a cyclic GMP analogue
The NO donor SNP (10 μm) enhanced HR response to 3 Hz (-EX[SNP]: from −62 ± 7 to −79 ± 9 beats min−1; +EX[SNP]: from −76 ± 8 to −98 ± 10 beats min−1; P < 0.01, unpaired t test) and 5 Hz VNS in -EX and +EX (P < 0.05, ANOVA; see Fig. 6), but responses remained enhanced in +EX compared to -EX (P < 0.05, ANOVA).
Figure 6. In vitro HR changes in response to VNS following administration of an NO donor, a sGC inhibitor and a cGMP analogue.
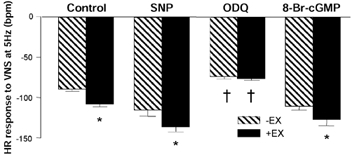
Heart rate (HR) changes to vagal nerve stimulation (VNS) at 5 Hz were significantly elevated in +EX compared to -EX (P < 0.05, unpaired t test). Responses were enhanced following administration of the nitric oxide (NO) donor sodium nitroprusside (SNP, 10 μm). However, the difference between -EX and +EX groups remained (P < 0.05, unpaired t test). Responses were attenuated following administration of the soluble guanylyl cyclase (sGC) inhibitor 1H-[1, 2, 4]oxadiazolo[4, 3-a]quinoxalin-1-one (ODQ) in both -EX and +EX groups (P < 0.01, ANOVA), and this drug also abolished the difference between groups. However, the cyclic guanosine monophosphate (cGMP) analogue 8-Br-cGMP (0.5 mm) increased the response to VNS (P < 0.05, ANOVA) in both -EX and +EX groups.
Inhibition of soluble guanylyl cyclase (sGC) with ODQ had the same effect as l-VNIO in +EX and -EX atria (3 Hz: -EX[ODQ]: from −62 ± 7 to −42 ± 8 beats min−1; +EX[ODQ]: from −76 ± 8 to −44 ± 8 beats min−1; 5 Hz: see Fig. 6; P < 0.05, ANOVA) and did not alter basal heart rate.
The cGMP analogue 8-Br-cGMP (0.5 mm) had a similar effect to SNP in both -EX and +EX atria (3 Hz: -EX[8-Br-cGMP]: from −62 ± 7 to −75 ± 4 beats min−1; +EX[8-Br-cGMP]: from −76 ± 8 to −86 ± 5 beats min−1; 5 Hz: see Fig. 6; P < 0.05, ANOVA), suggesting that downstream NO signalling was unaffected by training. Neither SNP, nor ODQ nor 8-Br-cGMP had any effect on the HR response to CCh (See Fig. 7), suggesting that NO-cGMP does not significantly modulate postsynaptic cholinergic signals regulating pacemaking.
Figure 7. In vitro HR changes in response to CCh following administration of an NO donor, a sGC inhibitor and a cGMP analogue.
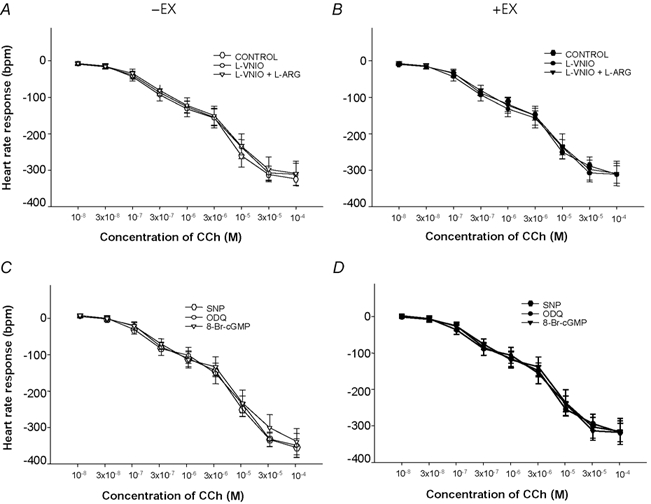
Heart rate (HR) changes in response to doses of carbamylcholine chloride (CCh) were not affected by NOS-1 inhibition with l-VNIO (100 μm; A and B) or a NO donor (sodium nitroprusside, 10 μm), a soluble guanylyl cyclase inhibitor (1H-[1, 2, 4]oxadiazolo[4, 3-a]quinoxalin-1-one (ODQ), 10 μm) or a cGMP analogue (8-Br-cGMP, 0.5 mm) (C and D).
SNP and 8-Br-cGMP both significantly increased basal HR (SNP: -EX, 355 ± 8 to 377 ± 9 beats min−1; +EX, 324 ± 6 to 347 ± 10 beats min−1; 8-Br-cGMP: -EX, 351 ± 9 to 367 ± 10 beats min−1; +EX 320 ± 6 to 337 ± 9 beats min−1; P < 0.01, unpaired t test). Both SNP and 8-Br-cGMP increase HR response to VNS independently of this basal HR change since blocking baseline responses (with an If inhibitor) does not affect the NO-cGMP response on cholinergic transmission or vagal bradycardia (Sears et al. 1999; Herring & Paterson, 2001).
Discussion
Here we show for the first time that the functional changes in cardiac vagal responsiveness associated with exercise training result from upregulation of NOS-1 in intracardiac cholinergic ganglia.
Beneficial effects of exercise training on the heart
The positive prognostic factors associated with regular exercise are numerous, and evidence produced over the past 50 years supports the concept that low physical activity is associated with increased mortality from a wide range of causes.
Periods of aerobic exercise training reduce resting HR (Yamamoto et al. 2001) and shorten the HR recovery phase following exercise (Imai et al. 1994). These HR changes are thought to be a result of increased vagal tone (Yamamoto et al. 2001) and rapid vagal reactivation at the cessation of physical activity (Sugawara et al. 2001). Furthermore, indices of parasympathetic activity such as accelerated recovery of heart rate following exertion (Imai et al. 1994) and high heart rate variability (Kleiger et al. 1987) have been linked to athleticism and emerged as strong independent correlates against sudden cardiac death (Hull et al. 1990; Cole et al. 1999). Although the extent to which impaired parasympathetic nerve activity is causal in the development of disease is not entirely clear, direct vagal nerve stimulation with chronically implanted devices has a powerfully antifibrillatory effect in dogs following prior myocardial infarctions (Vanoli et al. 1991).
Training upregulates NOS expression
Training enhances the bioavailability of NO in a range of tissues in the cardiovascular system. NOS-3-derived NO production is increased in the vasculature of skeletal muscle (Koller et al. 1995) and in the coronary circulation (Sessa et al. 1994). Conversely, impaired NO signalling is now a recognized feature of a number of vascular (Taddei et al. 2001) and myocardial diseases (Katz et al. 1993). Training also upregulates NOS-1 in cardiac sympathetic ganglia (Mohan et al. 2000) and, as shown here, in intracardiac parasympathetic ganglia. The mechanisms bringing about changes in NOS-1 expression following training are less well understood than those governing expression of the NOS-3 gene which contains a haemodynamic shear-stress responsive element (Ziegler et al. 1998). NOS-1 expression is responsive to physiological stress and in particular, cellular injury. Hypoxia (Guo et al. 1997) and pain (Lam et al. 1996) have both been shown to increase NOS-1 mRNA in neuronal tissues. However, given our present understanding of NOS-1 gene regulation, it is difficult to envisage a simple chain of events triggered by regular voluntary aerobic exercise that results in upregulated NOS-1 expression. It also is not clear whether training promotes the differentiation of ganglionic neurones into nitrergic neurones, or if it merely upregulates the expression of NOS-1 in cells already expressing this. There was a trend in our data showing an increased number of NOS-1-positive cell bodies relative to the ChAT-positive number following exercise training, but this failed to reach significance.
Isoform and site-specific action of NOS in modulating cardiac excitability
The site-specific function of NOS-1 is ultimately dependent on its cellular microdomain (Barouch et al. 2002). In particular, regulation of cardiac excitability by NOS-1 is almost certainly dependent on its location in one of two cellular compartments. First, NOS-1 localizes to the sarcoplasmic reticulum (SR) of ventricular myocytes (Xu et al. 1999), where it may act to regulate β-adrenergically enhanced calcium fluxes and myocardial contractility (Ashley et al. 2002; Barouch et al. 2002). Secondly, we and others have demonstrated a significant role for neuronal NO - derived from cholinergic ganglia (Choate et al. 2001) - in facilitating acetylcholine release (Herring & Paterson 2001) and vagal bradycardia in vitro (Herring et al. 2000) and in vivo (Conlon & Kidd, 1999; Markos et al. 2001). Furthermore, murine atrial NOS-1 staining was only evident at significant levels in cholinergic ganglia, suggesting that this NOS-1 may be quantitatively more important in the vagal control of heart rate. It is not entirely clear, however, whether the main functional role of ganglionic NOS-1-derived NO is facilitation of neurotransmission at cholinergic synapses within the ganglia, as some believe for the dog (Markos et al. 2002), or whether NO works predominantly at the neuroeffector junction, since we have observed that bradycardia induced by nicotinic receptor stimulation of post-ganglionic vagal neurones is facilitated by NO (E. J. F. Danson & D. J. Paterson, unpublished observation). However, given the increasingly diverse set of neuronal cell types and circuitry uncovered within cardiac parasympathetic ganglia (Edwards et al. 1995), it is not inconceivable that NO has more than one site of action in this region, which may be species dependent.
NOS-3 resident in a membrane caveolar matrix microdomain in atrial cells appears to have little role in the regulation of heart rate (Vandecasteele et al. 1999). It may, however, modulate basal and β-adrenergically stimulated contractile performance when NOS-3 protein levels are elevated as a result of transgenic over-expression (Brunner et al. 2002). This is consistent with our observation that inhibition of NOS had no effect on the intrinsic rate response to CCh before or after training and supports the idea that the main functional action of NO resides presynaptic to the cardiac myocyte.
Our data provide novel evidence that NO-dependent facilitation of vagal-induced bradycardia is augmented following exercise training. In vivo gene transfer of NOS-1 into the atrial wall mimics the exercise-trained vagal phenotype (Mohan et al. 2002), suggesting that NOS-1 is a key protein for increasing cardiac vagal gain of function.
Acknowledgments
We are grateful to the British Heart Foundation (BHF) for financial support and Dr R. M. Mohan for help and advice with the training regime. E.J.F.D. is a James Knott Research Scholar (St Cross College, Oxford).
References
- Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011–3016. doi: 10.1161/01.cir.0000019516.31040.2d. [DOI] [PubMed] [Google Scholar]
- Babu BR, Griffith OW. N5-(1-Imino-3-butenyl)-l-ornithine. A neuronal isoform selective mechanism-based inactivator of nitric oxide synthase. J Biol Chem. 1998;273:8882–8889. doi: 10.1074/jbc.273.15.8882. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104:3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- Buch AN, Coote JH, Townend JN. Mortality, cardiac vagal control and physical training - what's the link? Exp Physiol. 2002;87:423–435. doi: 10.1111/j.1469-445x.2002.tb00055.x. [DOI] [PubMed] [Google Scholar]
- Choate JK, Danson EJ, Morris JF, Paterson DJ. Peripheral vagal control of heart rate is impaired in neuronal NOS knockout mice. Am J Physiol - Heart and Circ Physiol. 2001;281:H2310–2317. doi: 10.1152/ajpheart.2001.281.6.H2310. [DOI] [PubMed] [Google Scholar]
- Cole CR, Blackstone EH, Pashkow FJ, Snader CE, Lauer MS. Heart-rate recovery immediately after exercise as a predictor of mortality. N Engl J Med. 1999;341:1351–1357. doi: 10.1056/NEJM199910283411804. [DOI] [PubMed] [Google Scholar]
- Conlon K, Kidd C. Neuronal nitric oxide facilitates vagal chronotropic and dromotropic actions on the heart. J Auton Nerv Syst. 1999;75:136–146. doi: 10.1016/s0165-1838(98)00185-4. [DOI] [PubMed] [Google Scholar]
- Edwards FR, Hirst GD, Klemm MF, Steele PA. Different types of ganglion cell in the cardiac plexus of guinea-pigs. J Physiol. 1995;486:453–471. doi: 10.1113/jphysiol.1995.sp020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvan A, Rubart M, Zipes DP. NO modulates autonomic effects on sinus discharge rate and AV nodal conduction in open-chest dogs. Am J Physiol. 1997;272:H263–271. doi: 10.1152/ajpheart.1997.272.1.H263. [DOI] [PubMed] [Google Scholar]
- Guo Y, Ward ME, Beasjours S, Mori M, Hussain SN. Regulation of cerebellar nitric oxide production in response to prolonged in vivo hypoxia. J Neurosci Res. 1997;49:89–97. [PubMed] [Google Scholar]
- Herring N, Golding S, Paterson DJ. Pre-synaptic NO-cGMP pathway modulates vagal control of heart rate in isolated adult guinea pig atria. J Mol Cell Cardiol. 2000;32:1795–1804. doi: 10.1006/jmcc.2000.1214. [DOI] [PubMed] [Google Scholar]
- Herring N, Paterson DJ. Nitric oxide-cGMP pathway facilitates acetylcholine release and bradycardia during vagal nerve stimulation in the guinea-pig in vitro. J Physiol. 2001;535:507–518. doi: 10.1111/j.1469-7793.2001.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull SS, Evans AR, Vanoli E, Adamson PB, Stramba-Badiale M, Albert DE, Foreman RD, Schwartz PJ. Heart rate variability before and after myocardial infarction in conscious dogs at high and low risk of sudden death. J Am Coll Cardiol. 1990;16:978–985. doi: 10.1016/s0735-1097(10)80351-1. [DOI] [PubMed] [Google Scholar]
- Imai K, Sato H, Hori M, Kusuoka H, Ozaki H, Yokoyama H, Takeda H, Inoue M, Kamada T. Vagally mediated heart rate recovery after exercise is accelerated in athletes but blunted in patients with chronic heart failure. J Am Coll Cardiol. 1994;24:1529–1535. doi: 10.1016/0735-1097(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Katz SD, Schwarz M, Yuen J, Lejemtel TH. Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure. Role of endothelium-derived vasodilating and vasoconstricting factors. Circulation. 1993;88:55–61. doi: 10.1161/01.cir.88.1.55. [DOI] [PubMed] [Google Scholar]
- Kleiger RE, Miller JP, Bigger JT, Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol. 1987;59:256–262. doi: 10.1016/0002-9149(87)90795-8. [DOI] [PubMed] [Google Scholar]
- Koller A, Huang A, Sun D, Kaley G. Exercise training augments flow-dependent dilation in rat skeletal muscle arterioles. Role of endothelial nitric oxide and prostaglandins. Circ Res. 1995;76:544–550. doi: 10.1161/01.res.76.4.544. [DOI] [PubMed] [Google Scholar]
- Lam HH, Hanley DF, Trapp BD, Saito S, Raja S, Dawson TM, Yamaguchi H. Induction of spinal cord neuronal nitric oxide synthase (NOS) after formalin injection in the rat hind paw. Neurosci Lett. 1996;210:201–204. doi: 10.1016/0304-3940(96)12702-6. [DOI] [PubMed] [Google Scholar]
- Markos F, Snow HM, Kidd C, Conlon K. Inhibition of neuronal nitric oxide reduces heart rate variability in the anaesthetised dog. Exp Physiol. 2001;86:539–541. doi: 10.1113/eph8602257. [DOI] [PubMed] [Google Scholar]
- Markos F, Snow HM, Kidd C, Conlon K. Nitric oxide facilitates vagal control of heart rate via actions in the cardiac parasympathetic ganglia of the anaesthetised dog. Exp Physiol. 2002;87:49–52. doi: 10.1113/eph8702303. [DOI] [PubMed] [Google Scholar]
- Mohan RM, Choate JK, Golding S, Herring N, Casadei B, Paterson DJ. Peripheral pre-synaptic pathway reduces the heart rate response to sympathetic activation following exercise training: role of NO. Cardiovasc Res. 2000;47:90–98. doi: 10.1016/s0008-6363(00)00066-3. [DOI] [PubMed] [Google Scholar]
- Mohan RM, Heaton DA, Danson EJF, Kinshaw SPR, Cai S, Channon KM, Paterson DJ. Neuronal nitric oxide synthase gene transfer promotes cardiac vagal gain of function. Circ Res. 2002 doi: 10.1161/01.res.0000047531.75030.b5. in the Press. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Vallance P, Amirmansour C, Harding SE. Positive inotropic effects of NO donors in isolated guinea-pig and human cardiomyocytes independent of NO species and cyclic nucleotides. Cardiovasc Res. 2000;48:430–439. doi: 10.1016/s0008-6363(00)00202-9. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK, Paterson DJ. Inhibition of nitric oxide synthase slows heart rate recovery from cholinergic activation. J Appl Physiol. 1998;84:1596–1603. doi: 10.1152/jappl.1998.84.5.1596. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK, Paterson DJ. NO-cGMP pathway accentuates the decrease in heart rate caused by cardiac vagal nerve stimulation. J Appl Physiol. 1999;86:510–516. doi: 10.1152/jappl.1999.86.2.510. [DOI] [PubMed] [Google Scholar]
- Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res. 1994;74:349–353. doi: 10.1161/01.res.74.2.349. [DOI] [PubMed] [Google Scholar]
- Sugawara J, Murakami H, Maeda S, Kuno S, Matsuda M. Change in post-exercise vagal reactivation with exercise training and detraining in young men. Eur J Appl Physiol. 2001;85:259–263. doi: 10.1007/s004210100443. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Sudano I, Salvetti A. Endothelial dysfunction in hypertension. J Cardiovasc Pharmacol. 2001;38:S11–14. doi: 10.1097/00005344-200111002-00004. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nat Med. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- Vanoli E, De Ferrari GM, Stramba-Badiale M, Hull SS, Foreman RD, Schwartz PJ. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. 1991;68:1471–1481. doi: 10.1161/01.res.68.5.1471. [DOI] [PubMed] [Google Scholar]
- Wang H, Morris JF. Presence of neuronal nitric oxide synthase in the suprachiasmatic nuclei of mouse and rat. Neuroscience. 1996;74:1059–1068. doi: 10.1016/0306-4522(96)00165-0. [DOI] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Miyachi M, Saitoh T, Yoshioka A, Onodera S. Effects of endurance training on resting and post-exercise cardiac autonomic control. Med Sci Sports Exerc. 2001;33:1496–1502. doi: 10.1097/00005768-200109000-00012. [DOI] [PubMed] [Google Scholar]
- Ziegler T, Silacci P, Harrison VJ, Hayoz D. Nitric oxide synthase expression in endothelial cells exposed to mechanical forces. Hypertension. 1998;32:351–355. doi: 10.1161/01.hyp.32.2.351. [DOI] [PubMed] [Google Scholar]