

Abstract
The modulatory role of endothelial nitric oxide synthase (eNOS) on heart contraction, relaxation and rate is examined in light of recent studies using genetic deletion or overexpression in mice under specific conditions. Unstressed eNOS-/- hearts in basal conditions exhibit a normal inotropic and lusitropic function, with either decreased or unchanged heart rate. Under stimulation with catecholamines, eNOS-/- mice predominantly show a potentiation in their β-adrenergic inotropic and lusitropic responsiveness. A similar phenotype is observed in β3-adrenoceptor deficient mice, pointing to a key role of this receptor subtype for eNOS coupling. The effect of eNOS on the muscarinic cholinergic modulation of cardiac function probably operates in conjunction with other NO-independent mechanisms, the persistence of which may explain the apparent dispensability of this isoform for the effect of acetylcholine in some eNOS-/- mouse strains. eNOS-/- hearts submitted to short term ischaemia-reperfusion exhibit variable alterations in systolic and diastolic function and infarct size, while those submitted to myocardial infarction present a worsened ventricular remodelling, increased 1 month mortality and loss of benefit from ACE inhibitor or angiotensin II type I receptor antagonist therapy. Although non-conditional eNOS gene deletion may engender phenotypic adaptations (e.g. ventricular hypertrophy resulting from chronic hypertension, or upregulation of the other NOS isoforms) potentially confounding the interpretation of comparative studies, the use of eNOS-/- mice has undoubtedly advanced (and will probably continue to improve) our understanding of the complex role of eNOS (in conjunction with the other NOSs) in the regulation of cardiac function. The challenge is now to confirm the emerging paradigms in human cardiac physiology and hopefully translate them into therapy.
Although the enzymatic capacity of endothelial cells to produce nitric oxide was described as early as 1987 (Palmer et al. 1987), the definitive molecular characterization of the endothelial isoform of nitric oxide synthase, or eNOS, was first provided in 1992 (Janssens et al. 1992; Lamas et al. 1992; Nishida et al. 1992; Sessa et al. 1992). Analysis of its cDNA confirmed its belonging to a family of three different NO synthases, each encoded by a different gene, i.e. the neural isoform (or nNOS), the inducible isoform (or iNOS, initially cloned from monocytes/macrophages) and the endothelial isoform (eNOS), encoded by NOS1, NOS2 and NOS3, respectively. Subsequent expressional studies revealed each isoform to be present in many more cell types than where it was originally discovered, both at the mRNA and at the protein level. Accordingly, eNOS was identified in hippocampal neurons, several epithelial cell types, platelets and cardiac myocytes, in addition to endothelial cells (for a review, see (Forstermann et al. 1998). Not unexpectedly, this promiscuity also results in the co-expression of several NOS isoforms within the same cell type, as exemplified in cardiac muscle cells where the constitutive eNOS and nNOS may coexist with iNOS, induced upon stimulation with the appropriate inflammatory mediators. A mitochondrial NOS (mtNOS) has also been identified (Bates et al. 1996), corresponding to a variant of neuronal NOS (Kanai et al. 2001; Elfering et al. 2002), although its functional impact on mitochondrial function in vivo remains to be firmly established (French et al. 2001).
Despite this apparent redundancy, the use of isoform-specific inhibitors or genetic deletion experiments has identified a specific modulatory role for each isoform that is subserved by its subcellular localization, at least in the cardiomyocyte. The recent publication of the phenotypic characterization of genetically modified mice in which eNOS is either deleted or overexpressed specifically in cardiomyocytes now motivates the proposal of an updated model of its regulatory functions, almost 10 years after the initial description of the modulation of catecholamine responsiveness of isolated cardiac cells by endogenous NO (Balligand et al. 1993).
Molecular regulation of eNOS: an update
Like the other two members of the NOS enzyme family, eNOS contains two functionally distinct domains, i.e. an N-terminal oxygenase, where haeme, tetrahydrobiopterin (BH4) and l-arginine bind, and a C-terminal reductase comprising binding sites for FAD, FMN and NADPH. These two domains are linked by a calmodulin-binding site (aa 499 to 518 of human eNOS) where, upon calcium-induced binding, calmodulin increases the rate of electron transfer from NADPH to the reductase domain flavins and from the reductase domain to the haeme centre for the oxidation of the substrate, l-arginine. Accordingly, inhibition of eNOS activity by calcium removal and calmodulin inhibitors justifies the classification of eNOS (with nNOS) as one of two calcium-sensitive NOSs. Moreover, all NOS function in a dimeric form that is stabilized by haeme and l-arginine, as well as BH4 (Marletta, 1993; Crane et al. 1998). Importantly, in the absence of sufficient l-arginine or BH4, ‘uncoupled’ NOS may generate superoxide anions (O2−) instead of NO (Pou et al. 1992), leading to the formation of peroxynitrite (ONOO−) resulting from the equimolar reaction of NO and O2−. In turn, peroxinitrite may further induce the production of O2− by eNOS through oxidation of its zinc-sulphur cluster (Zou et al. 2002). The N-terminal domain also contains a glycine residue in position 2 critical for myristoylation, as well as cysteines (Cys15 and Cys26) supporting palmitoylation that are unique to eNOS and importantly condition the enzyme targeting to caveolae (Feron et al. 1998a). Caveolae are small (70-90 nm in diameter), invaginated foldings of the plasmalemmal membrane that are distinctively enriched in cholesterol and glycosphingolipids, and structurally maintained by oligomerized caveolins that also serve as scaffolds for the assembly of multi-protein signalling complexes within these specialized membrane compartments (Simons & Toomre, 2000). Of note, T-tubule membranes are particularly enriched in caveolae (Levin & Page, 1980) which supports a modulatory role of eNOS in excitation-contraction coupling in the sarcoplasmic reticulum (SR)-T-tubule junctional space (e.g. in response to stretch; Petroff et al. 2001), where it may act in concert with nNOS also expressed in SR membranes (Xu et al. 1999).
Aside from transcriptional up- and down-regulation (for a review, see Forstermann et al. 1998) and the availability of cofactors and substrate (which support eNOS dimeric conformation for optimal catalytic activity), eNOS is also regulated post-translationally by phosphorylation on serine, and, in specific circumstances, also on tyrosine and threonine residues. Stimuli such as insulin or stretch (in cardiomyocytes) (Petroff et al. 2001) induce phosphorylation at serine 1177 (for human)/1179 (for bovine eNOS) through PI3K-dependent activation of Akt (protein kinase B), with a subsequent increase in enzyme activity that is less sensitive to fluctuations of intracellular calcium through mechanisms that are presently unclear. Other kinases, e.g. protein kinase A, protein kinase G and AMP-activated kinase also phosphorylate eNOS on serine 1177. AMP-activated kinase and protein kinase C also induce phosphorylation on threonine 495 that inactivates eNOS. According to current models in endothelial cells, stimulation with histamine or bradykinin would first lead to dephosphorylation at this threonine residue, located in the critical calmodulin binding domain, to allow the binding of calmodulin, further stabilized by the subsequent phosphorylation on serine 1177 (Fleming et al. 2001).
Regulation of eNOS by protein-protein interactions
Caveolin
eNOS interacts with caveolin-1 (and caveolin-3) in assays of recombinant proteins in vitro or in co-immunoprecipitation assays using anti-caveolin-1 antibodies from endothelial cell extracts (and anti-caveolin-3 antibodies in extracts of cardiomyocytes). This interaction both ensures the proper targeting of eNOS (or at least a portion of cellular eNOS) to caveolae and maintains eNOS in an inhibited state. This inhibition can be reversed by addition of exogenous calmodulin, suggesting a reciprocal regulation of the enzyme by inhibitory caveolin versus activating calcium-calmodulin (Michel et al. 1997). A current model proposes that stimulus- or agonist-induced increases in intracellular calcium promote the displacement of inhibitory caveolin and binding of activated calcium- calmodulin to its consensus sequence on eNOS to initiate catalytic activity. Whether this necessarily implicates translocation of eNOS out of caveolae is still under debate. The phenotypes of mice deficient in either caveolin-1 or caveolin-3 illustrate the functional relevance of the inhibitory ‘caveolin clamp’ in vivo, e.g. vessels from caveolin-1 deficient mice exhibit a marked hyporesponsiveness to constrictor agonists attributable to increased NO release (Drab et al. 2001; Razani et al. 2001) whereas increased nNOS activity has been observed in skeletal muscle from caveolin-3 deficient mice (Sunada et al. 2001) Likewise, our group showed that statins potentiate eNOS activity by decreasing caveolin-1 abundance in vitro and in vivo, at least in macrovascular endothelial cells where the caveolin pool is lower and the proportion of caveolin-bound eNOS is higher (Feron et al. 2001).
The allosteric regulation of eNOS could theoretically be influenced upon stoichiometric changes in the abundance of any of its protein partners that would impact on its binding equilibrium. At one extreme is the total absence of a binding partner, as exemplified in genetic deletion experiments for caveolin-1 and caveolin-3, as mentioned above. Whether the abundance of these proteins changes with pathologic states, especially in heart diseases, has been very little explored. Recently, we showed a reduction of caveolin-1 and −3 (and eNOS) protein abundance in left ventricular tissue of dogs with non-failing, hypertrophic cardiomyopathy induced by perinephritic hypertension (Piech et al. 2002b) and in similar extracts from spontaneously hypertensive rats (SHR) (Piech et al. 2002a). Of note, despite reduced eNOS abundance, the tissue levels of cGMP were unchanged, and these animals retained a marked sensitivity to NOS inhibitors, indicating that eNOS catalytic activity was probably maintained through the parallel downregulation of inhibitory caveolins. This emphasizes the need to integrate changes in NOS abundance with those of their allosteric regulators in future studies on cardiovascular diseases to gain further understanding in their functional impact on downstream NO signalling.
Hsp 90
This ubiquitous 90 kDa, heat-shock protein is expressed at high levels (accounting for up to 1-2 % of total cellular protein content) in the cytosol even in unstressed conditions. It functions as a chaperone for the proper folding of specific protein substrates, including many signal transducing molecules (e.g. non-receptor tyrosine kinases, transcription factors and eNOS, among others; for a review, see Richter & Buchner, 2001). Most of its regulatory action in eNOS signalling has been described in endothelial cells. Hsp90 is associated with eNOS in resting endothelial cells and, upon stimulation with vascular endothelial growth factor (VEGF), oestrogen, histamine, shear stress and statins, the association between the two proteins is increased, resulting in enhanced NO production (Garcia-Cardena et al. 1998). Of note, the protein kinase Akt (or protein kinase B), the kinase involved in the activating phosphorylation of eNOS on serine 1177, is another client protein for hsp90 and binds to a sequence of hsp90 that does not overlap with those involved in the binding of eNOS. Therefore, hsp90 was recently proposed as an adaptor between Akt and its substrate, eNOS, thereby promoting the activating phosphorylation of eNOS (for more details, see Balligand, 2002).
Modulation of cardiac function by eNOS
We will focus on the specific impact of NO, as released from eNOS, on inotropic, lusitropic and chronotropic aspects of cardiac contraction (for a comprehensive review on all NOSs, see also Massion et al. 2001). This will encompass the paradigms deduced from studies in isolated cardiomyocytes, isolated cardiac muscle preparations and in vivo assessment of cardiac haemodynamics in animals (mostly in mice with genetic deletion of eNOS, as mentioned in the Introduction), and to a limited extent, in humans. We will distinguish the influence of eNOS in unstimulated cardiac preparations (i.e. in the basal state, without agonist activation) and in stimulated ones (e.g. with catecholamines). In addition, the role of eNOS in unstimulated and stimulated preparations will be compared in ‘unstressed’ (i.e. in the absence of cardiac injury, such as ischaemia-reperfusion, or infarction) and in ‘stressed’ preparations.
Role of eNOS in ‘unstressed’ and ‘basal’ cardiac preparations
Effect on basal contractility
Some studies previously reported a biphasic inotropic effect of exogenous NO, positive with low concentrations of NO donors but negative with high ones (Kojda et al. 1996; Mohan et al. 1996; Vila-Petroff et al. 1999; Wegener et al. 2002). Endogenous NO also may have a positive inotropic effect (Kojda et al. 1997; Muller-Strahl et al. 2000), as recently evidenced in normal human hearts (Cotton et al. 2001). A positive inotropic effect of NO may be explained at the cardiomyocyte level by the potential following mechanisms (see Fig. 1): (1) direct activating nitrosylation of the RyR2 (Xu et al. 1998), as demonstrated by nanomolar NO in skeletal muscle (RyR1) (Eu et al. 2000) and also probably accounting for the enhanced excitation- contraction coupling gain and positive inotropic effect of cardiomyocyte stretch (Petroff et al. 2001); (2) direct S-nitrosylation of the voltage-operated L-type calcium channel (VOC) through a redox switch-mediated increase in ICa,L (Campbell et al. 1996); (3) cGMP-independent activation of adenylyl cyclase at low NO levels (Vila-Petroff et al. 1999); (4) cGMP-dependent increase in cAMP, through cGMP-mediated inhibition of cAMP PDE III and prevention of cAMP breakdown (Mery et al. 1993); (5) PKG-mediated activation of the RyR, through phosphorylation of ADP ribosyl cyclase and cADP-ribose-mediated activation of RyR, as identified in sea urchin eggs (Willmott et al. 1996). However, one should bear in mind that the above mechanisms were mostly demonstrated with exogenous NO donors that may not always reproduce the action of endogenous NO released within the boundaries of cellular microdomains. Accordingly, eNOS gene deletion does not influence basal cardiac inotropic state in mice (see Table 1), although this apparently ‘neutral’ phenotype may result from the confounding effects of several compensatory adaptations. These may include (1) ‘backup’ production of NO by nNOS, recently shown to exert autocrine modulation of cardiomyocyte function (albeit in divergent fashion - see Barouch et al. (2002) and Ashley et al. (2002); (2) or iNOS, superinduced in eNOS-/- mice even in unstressed conditions (Sharp et al. 2002); (3) production of atrial natriuretic peptide (Gyurko et al. 2000). The latter would increase myocardial cGMP, with effects mostly on relaxation, rather than inotropism (Ji et al. 1999). In addition, chronic hypertension in older eNOS-/- mice may induce structural adaptations characteristic of the hypertrophic phenotype, such as reduced arteriolar density (Kubis et al. 2002), altered SR calcium load (Boknik et al. 2001) or expression of ionic channels (e.g. an increase in IK,ACh (Guo et al. 1997), all of which may confound the interpretation of comparative studies with wild-type animals. Conversely, transgenic mice with overexpression of the eNOS gene driven by a cardiomyocyte-specific promoter exhibited a reduction in basal contractile state, probably attributable to the very high level of eNOS expression and deregulated NO production (90-fold higher basal eNOS activity) (Brunner et al. 2001). The negative inotropic effect in this case probably involves a desensitization of cardiac contractile myofilaments to calcium.
Figure 1. NO signalling pathways in the cardiomyocyte.
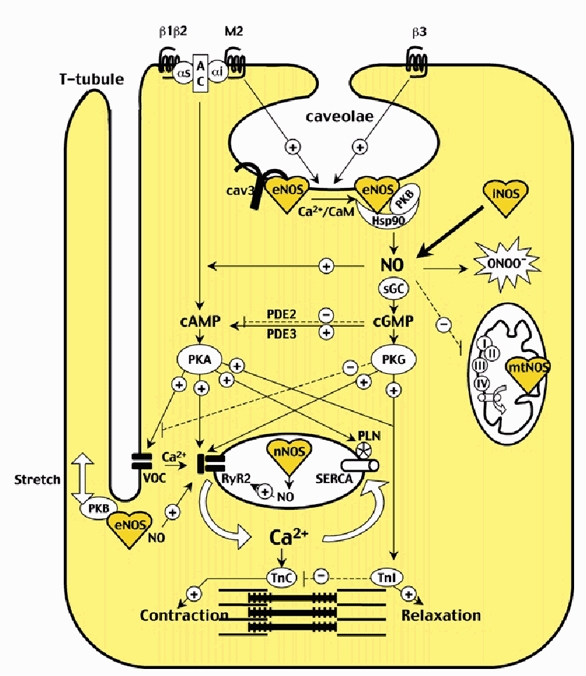
All NOS isoforms (eNOS, nNOS, iNOS and mtNOS) within specific subcellular compartments of the cardiomyocyte (e.g. sarcolemmal caveolae, SR-T-tubule junction, sarcoplasmic reticulum, mitochondria) are represented, as well as associated regulatory proteins. The net effect of eNOS-derived NO on contractility depends on the specific stimulus (e.g. agonists on β3-adrenoceptor and muscarinic M2 receptor, or stretch), the subcellular compartment (cytosolic or subsarcolemmal) involved and the subsequent amount of NO produced as well as the oxidative status of the cell. On one hand, NO exerts anti-adrenergic inotropic effects after β3-adrenoceptor-dependent activation of eNOS and through cytosolic (and mostly cGMP-dependent) modulation of the main targets of the classical β-adrenergic cAMP-protein kinase A (PKA) pathway, i.e. (1) the voltage-operated L-type Ca2+ channel (VOC); (2) the ryanodine receptor Ca2+-release channel type 2 (RyR2); (3) phospholamban (PLN; involved in regulation of sarcoendoplasmic reticulum Ca2+-ATPase (SERCA)); (4) the troponin I (TnI) limiting the sensitivity of troponin C (TnC) to Ca2+. On the other hand, eNOS-derived NO may exert positive inotropic effects, e.g. (1) after sarcolemmal stretch, through subsarcolemmal activation of protein kinase B (PKB), eNOS and probably direct nitrosylation of RyR2 by NO; (2) direct nitrosylation of VOC; (3) direct activation of adenylate cyclase (AC); (4) increase of cAMP after phosphodiesterase 3 (PDE3) inhibition; (5) activation of RyR2 through protein kinase G (PKG)-dependent ADP ribosyl cyclase phosphorylation. αs and αi, G protein subtypes; β1, β2 and β3, adrenoreceptor subtypes; cav3, caveolin 3; CaM, calmodulin; ONOO−, peroxynitrite; I to IV, mitochondrial respiratory complexes; full arrows with + symbol designate stimulation; dashed arrows with - symbol designate inhibition.
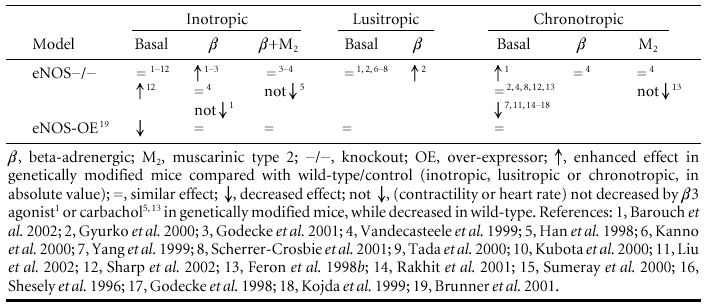
Table 1.
Inotropic, lusitropic and chronotropic effects of NO in unstressed hearts, under basal and β-adrenergic- and/or muscarinic-stimulated conditions
Effect on basal relaxation
NO exerts a positive lusitropic effect that has been attributed to a cGMP- and PKG-mediated phosphorylation of troponin I, subsequent myofilament calcium desensitization, relaxation hastening and improved distensibility (Shah et al. 1994; Layland et al. 2002). Indeed, previous studies showed that infusion of intracoronary NO donors in patients induced (1) a relaxation-hastening effect (shorter time to onset of left ventricular (LV) relaxation), secondary (or leading) to abbreviation of contraction (associated with reduced LV end systolic pressure); (2) an improved LV diastolic distensibility (downward shift of the end diastolic pressure-volume relationship, i.e. greater LV end diastolic volume at lower LV end diastolic pressure) (Paulus et al. 1994, 1995; Bartunek et al. 1997; Paulus, 2001), especially in hypertrophied hearts (Matter et al. 1999). The velocity of relaxation (-dP/dtmin) was not improved in these patients (Paulus et al. 1994), contrary to some animal models (Muller-Strahl et al. 2000; Hart et al. 2001). Notably, the above effects were observed either with exogenous NO donors (in vivo, or on isolated cardiomyocytes) or after stimulation with agonists known to activate paracrine NO production from endothelial cells (in whole heart preparations), but the specific implication of unstimulated endothelial or cardiomyocyte eNOS remains uncertain. Analysis of the cardiac phenotype of genetically modified mice revealed that neither eNOS -/- nor cardiomyocyte eNOS-overexpressing mice presented any modification in relaxation under basal conditions in vivo (Table 1), which would suggest that eNOS is dispensable for normal diastolic function in vivo. The absence of eNOS-derived NO may be compensated by intracardiac production of atrial natriuretic peptide, another stimulant of guanylyl cyclase and cGMP, potentially leading to similar lusitropic properties (Gyurko et al. 2000), or NO from residual nNOS with similar positive lusitropic properties, as illustrated in a recent study on nNOS-/- mice (Ashley et al. 2002).
Effect on basal heart rate
A distinction must be made here between the involvement of eNOS on spontaneous rhythmicity of pacemaker cells versus the modulation by eNOS of the effect of autonomic (i.e. orthosympathetic and parasympathetic) agonists on the heart. Not considered here, but probably relevant to whole organ physiology, is the effect of endogenous NO (from nNOS as well as eNOS) on the control of neuromediator release at the pre-synaptic level, that negatively affects the orthosympathetic input, but reinforces the vagal influence to the heart (Elvan et al. 1997; Choate et al. 2001). At the cardiomyocyte level, cGMP analogues and NO from endogenous NOS were shown to decrease the spontaneous beating rate of cultured neonatal ventricular myocytes (Balligand et al. 1993). Feron et al. (1998b) subsequently showed the abolition of the response to carbachol in neonatal myocytes from eNOS-/- mice, although the generalizability of this NO dependence for the vagal effect in other cardiac preparations is subject to caution. In eNOS-deficient mice, baseline heart rate was increased in only one study (Barouch et al. 2002), possibly owing to a relative hypovolaemia and/or chronic ventricular remodelling, while the other studies showed normal or decreased heart rate. Since all conscious (unanaesthetised) 2- to 3-month-old eNOS-/- mice (Shesely et al. 1996; Kojda et al. 1999; Yang et al. 1999; Liu et al. 2002) presented a decreased heart rate, it is reasonable to assume a positive chronotropic effect of eNOS at the whole organ level. Low concentrations of NO donors were previously shown to increase heart rate in vitro (Pabla & Curtis, 1995; Musialek et al. 1997), possibly through stimulation of the hyperpolarization-activated inward current (If) sensitive to cGMP (Musialek et al. 1997, 2000; Yoo et al. 1998). The bradycardia observed in eNOS-/- mice may also result from a potential adaptive hyperactivity of nNOS, known to facilitate acetylcholine release at the presynaptic level (Jumrussirikul et al. 1998; Choate et al. 2001).
Role of eNOS in unstressed but stimulated hearts
Effect on stimulated contractility
The situation is quite different after activation of the β-adrenergic pathway. Under β-adrenergic stimulation in vivo, eNOS knockout mice (from two different strains) exhibited a potentiation of their inotropic response (Gyurko et al. 2000; Barouch et al. 2002). A similar observation was made in isolated mouse hearts from yet another strain, after perfusion with intracoronary dobutamine (Godecke et al. 2001). In papillary muscle preparations, another group did not find any difference in the inotropic response to isoproterenol (Vandecasteele et al. 1999). Although the apparent discrepancy between paradigms observed in whole organs versus papillary muscles may have suggested a significant contribution from paracrine (i.e. endothelial-derived NO) under flow conditions, when contractile shortening was studied in isolated, single cardiomyocytes from eNOS knockout mice, Barouch and colleagues again observed a potentiation of the effects of isoproterenol (Barouch et al. 2002), concomitant with an increased calcium transient. This phenotype implies a countervailing effect of autocrine, eNOS-derived NO on the inotropic response to β-adrenergic stimulation, as first suggested almost a decade ago (Balligand et al. 1993). Subsequent evidence has indeed demonstrated that β-adrenergic agonists activate a calcium-sensitive NOS in cardiomyocytes (Balligand et al. 1995; Kanai et al. 1997). Although the molecular mechanism for this activation is incompletely characterized, convergent evidence (Gauthier et al. 1998; Moniotte et al. 2001) clearly identified the involvement of β3-adrenoceptors for the stimulation of NO production in the human myocardium. Subsequent studies showed that the negative inotropic effect (and decrease in calcium transient) induced by the β3-preferential agonist BRL 37344 is abolished in cardiomyocytes from eNOS-/- mice (Barouch et al. 2002), and that the potentiation of the β-adrenergic inotropic effect of NO synthase inhibitors is absent in β3-adrenoceptor knockout mice (Varghese et al. 2000), thereby extending the validity of our paradigm in genetically deficient mice.
Once activated, eNOS may counterbalance the adrenergic effect by at least three pathways (see Fig. 1), some of which were identified on the basis of adrenergic inhibition with exogenous NO donors (Mery et al. 1993; Campbell et al. 1996; Vila-Petroff et al. 1999), e.g. (1) cGMP-dependent inhibition of the voltage-operated L-type calcium channel (VOC) and subsequent inhibition of ICa,L (Campbell et al. 1996), either by PKG-dependent phosphorylation of an intermediate protein opposing the effect of PKA (Mery et al. 1991) or by increasing cGMP-sensitive cAMP phosphodiesterase (PDE) II activity (Mery et al. 1993; Han et al. 1998); (2) PKG-dependent phosphorylation of troponin I decreasing troponin C-mediated myofilament responsiveness to calcium (Blumenthal et al. 1978; Lincoln & Corbin, 1978), as reproduced with a cGMP analogue (Shah et al. 1994), NO donors (Vila-Petroff et al. 1999) or NOS stimulation by pacing (Kaye et al. 1999); (3) inhibition of RyR2 by NO (Zahradnikova et al. 1997), at least after maximal β-adrenergic stimulation (Ziolo et al. 2001). Since β-adrenergic-stimulated ICa,L was unchanged in most experiments using cardiomyocytes from eNOS-/- mice (Vandecasteele et al. 1999; Belevych & Harvey, 2000; Godecke et al. 2001), the first of the above mechanisms seems unlikely, at least in mouse cardiomyocytes.
Aside from a reduced basal LV-developed pressure, as mentioned above, mice with a large amount of eNOS overexpression (40- to 90-fold, based on enzymatic activity; Brunner et al. 2001) exhibited a downward shift of the isoproterenol dose-response curve for left ventricular developed pressure (LVDP) but with unchanged EC50, suggesting that the β-adrenergic effect was superimposed on a constant background production of NO, as if the (vastly) overexpressed eNOS was uncoupled from agonist stimulation in this strain. In contrast, in another strain of transgenic mice with lower amounts of cardiomyocyte-specific eNOS overexpression, the baseline inotropic state of the heart, assessed from LV +dP/dtmaxin vivo, was unchanged, whereas the inotropic response to isoproterenol was attenuated at higher doses of the agonist (S. Janssens, personal communication).
In addition to modulating the positive inotropic effect of β-adrenergic agonists, eNOS was also suggested to mediate, at least in part, the attenuating effect of muscarinic cholinergic stimulation on the β-adrenergic response (Balligand et al. 1995; Han et al. 1995, 1996; Hare et al. 1995), i.e. the classical ‘accentuated antagonism’ (Levy, 1971). This proposition, however, has been challenged on the basis of subsequent negative studies (Vandecasteele et al. 1999; Belevych & Harvey, 2000; Godecke et al. 2001; Bett et al. 2002). By contrast, accentuated antagonism was lost in another study using eNOS-/- myocytes (Han et al. 1998). Clearly, eNOS is not an obligatory pathway in all species (notably not in frogs) and its implication relative to other signalling mechanisms (e.g. muscarinic cholinergic Gi/o coupling to IK-Ach or inhibition of adenylyl cyclase) varies at different levels of the heart, i.e. atria versus ventricles and pacemaking versus working myocytes, which may confound the interpretation of experiments using whole heart preparations. Since these alternative, eNOS-independent pathways (as well as other confounding NO-sensitive currents, such as If) are less represented at the ventricular level, this is where the modulatory role of eNOS may be more easily identifiable. Nevertheless, even when using ventricular myocytes, other technical parameters turned out to be crucial for the proper identification of eNOS influence, such as the temperature used for in vitro experiments, which is critical for enzymatic activity. Other factors (age-dependent presence of ventricular hypertrophy and potential upregulation of IK-Ach, non-littermate genetic background for control groups) have confounded the interpretation of experiments in eNOS-/- mice that were reported as negative (Vandecasteele et al. 1999). A more complete analysis of this contradictory evidence can be found in a previous review on the subject (Balligand, 1999).
Other stimuli than neurotransmitters or hormones may also activate eNOS in the cardiomyocyte. Sarcomere stretching (in the range of physiological elongation, i.e. 12-14 % increase in length) was recently shown to increase eNOS phosphorylation on Ser1179 (consecutive to PI3 kinase activation and phosphorylation of downstream Akt) and induce measurable increases in NO production in single myocytes. This effect was accompanied with an increase in calcium spark rate and a slow increase in calcium transients that was totally absent in cardiomyocytes from eNOS-deficient mice. Notably, these effects were also insensitive to guanylyl cyclase inhibition with1-H-[1,2,4]oxidiazolo[4,3-α]quinoxaline-1-one (ODQ), pointing to a cGMP-independent mechanism, possibly through S-nitrosylation of RyR2 (Petroff et al. 2001). The relative enrichment of T-tubules in caveolar membranes (where eNOS is localized) would favour a role for eNOS-derived NO in the regulation of EC coupling through its compartmentation in the SR-T-tubule junction, as opposed to its modulation of other aspects of cardiac contraction (as detailed above) that mostly involve increases in cytosolic cGMP. This eNOS-mediated increase in EC coupling gain with stretch could participate in the length-dependent recruitable contractile reserve capacity of the heart, accounting for at least part of the classical Anrep effect.
Effect on stimulated relaxation
Again, the situation may be different under β-adrenergic stimulation, that, by itself, induces a well-known positive lusitropic effect. As for inotropy (see above), β-adrenergic activation of eNOS may similarly oppose the effect of catecholamines on relaxation, instead of being additive. This would be supported by molecular data (Layland et al. 2002) showing that troponin I is phosphorylated on the same residues by PKG and PKA, and that the effect of 8-bromo-cGMP to desensitize cardiac myofilaments to calcium was abolished in the presence of isoproterenol (Shah et al. 1994) (again arguing for effects of the two interventions that are mutually exclusive). Accordingly, eNOS-/- mice had an improved relaxation under β-adrenergic stimulation (as attested by enhanced -dP/dtmin compared to wild-type littermate (Gyurko et al. 2000).
Effect on stimulated heart rate
eNOS-/- mice presented no difference in their increase in heart rate under β-adrenergic stimulation compared to controls (Vandecasteele et al. 1999), but more inducible ventricular tachycardia after digoxin pretreatment (Rakhit et al. 2001) as well as more ouabain-induced arrhythmic contractions and transient inward current (Kubota et al. 2000). NO has been shown to transiently increase (Herring et al. 2001) or decrease (Yoo et al. 1998) If during adrenergic stimulation in sinoatrial node pacemaker cells. These studies point to a potential role of eNOS in controlling the sensitivity to arrhythmia that deserves more study in the clinical setting.
Role of eNOS in the stressed heart
Although seminal studies have provided valuable insights into the role of NO in the stressed heart using NOS inhibitors, the relative lack of specificity of these drugs (towards the three NOS isoforms) precludes firm conclusions regarding the specific role of cardiac eNOS. Therefore, emphasis will be put on the latest studies of the phenotype of mice genetically deficient in eNOS submitted to various cardiac insults.
eNOS in acute ischaemia-reperfusion
During ischaemia- reperfusion (I/R), eNOS-/- mice exhibit either improved or decreased contractility, improved or worsened relaxation, unchanged heart rate as well as variable impact on infarct size (see Table 2). Differences in ischaemic (range 16-30 min) or reperfusion times (30-60 min) in the experimental control of coronary flow and heart rate may complicate the interpretation. Studies with the shortest ischaemic time (16 min) at constant coronary flow and pacing (600 min−1) (Flogel et al. 1999) showed an improved functional inotropic (LVP), lusitropic and metabolic (phosphocreatine and ATP) recovery, suggesting a detrimental effect of NO in I/R, at least in isolated hearts under these experimental conditions. Possibly, the endogenous production of NO combined with the classical oxidant burst of O2− in the initial phase of reperfusion may produce peroxynitrite, a well-known mediator of cellular injury (Beckman et al. 1990; Wang & Zweier, 1996; Yasmin et al. 1997). By contrast, another study (Sumeray et al. 2000) in eNOS-/- mice showed an increased infarct size attributable to a permanent decrease in coronary flow, confirming the protective vasodilatory properties of NO. Furthermore, endogenous NO produced by additional NOS isoform induction may exert cardioprotective effects. Accordingly, eNOS-/- mice in the study by Kanno et al. (2000) showed a compensatory induction of iNOS after 30 min of ischaemia that resulted in improved function attributed to cardioprotective iNOS-derived NO production. Finally, eNOS-/- mice also lost the benefit from preconditioning with repetitive ischaemic cycles (Bell & Yellon, 2001) and an ACE inhibitor (Yang et al. 1999), emphasizing the benefit of NO produced by vascular eNOS in these settings.
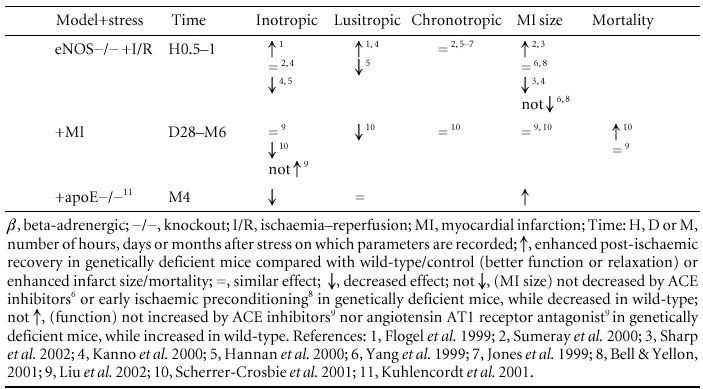
Table 2.
Inotropic, lusitropic and chronotropic effects of NO in stressed hearts, under basal conditions, with correspondant infarct size and mortalityspan
The picture may again be different when considering the role of eNOS in the late window of preconditioning (delayed acquisition of tolerance to ischaemia). A substantial body of evidence now supports a critical role for constitutive NOS in the early triggering (Xuan et al. 2000) of the induction of a second isoform, or iNOS, that, in turn, ensures a sustained production of cardioprotective NO. Specifically, enhanced NO production by iNOS, moderately and specifically overexpressed in myocytes (Wang et al. 2002), is essential to mediate the anti-stunning and anti-infarct actions of late preconditioning elicited by five different stimuli (ischaemia, adenosine A1 agonists, opioid δ1 agonists, endotoxin derivatives and exercise), suggesting that the upregulation of this enzyme is a central mechanism whereby the myocardium protects itself from ischaemia (Guo et al. 1999). The molecular and functional aspects of ischaemia-induced late preconditioning can be reproduced by the administration of NO donors in lieu of ischaemia in experimental animals and more recently in patients (Leesar et al. 2001), indicating that NO is also sufficient to induce late preconditioning. Accordingly, gene transfer of either eNOS or iNOS has been shown to replicate the infarct-sparing actions of ischaemic preconditioning, suggesting that NOS gene therapy could be an effective strategy for alleviating ischaemia-reperfusion injury (for a complete review, see Bolli, 2001).
eNOS in chronic myocardial infarction
When submitted to coronary ligation, eNOS-/- mice had unchanged infarct size but evident remodelling with decreased capillary density and hypertrophy (unrelated to the development of hypertension), accompanied with subsequent systolic and diastolic dysfunction and increased mortality at 28 days (Scherrer-Crosbie et al. 2001). This points to a beneficial effect of eNOS-derived NO on ventricular remodelling after myocardial infarction, as evidenced recently with the eNOS-mediated cardioprotective effects of corticoids (Hafezi-Moghadam et al. 2002; Thiemermann, 2002), possibly by increasing coronary collateralization and limiting myocyte hypertrophy. Double eNOS and apoE knockout mice with spontaneous coronary atherosclerosis revealed depressed contractility and increased infarct lesions at 4 months compared with apoE-/- mice (Kuhlencordt et al. 2001). A chronically reduced coronary reserve by eNOS ablation may again account for this result. In more chronic post-infarction remodelling conditions (6 months) (Liu et al. 2002), no difference has been found between eNOS-/- and wild-type, suggesting an adaptation through compensatory mechanisms (prostacyclin, nNOS, adaptation of remote myocardium), so that the influence of eNOS in the development and progression of chronic ischaemic heart failure may be considered less prominent, at least in the mouse model. However, the absence of eNOS significantly decreased the long term cardioprotective effects of an ACE inhibitor and an angiotensin II type I receptor antagonist (Yang et al. 1999; Liu et al. 2002), emphasizing the importance of eNOS-derived NO in modulating the growth/remodelling effects of mediators of the renin-angiotensin system.
Conclusion
The regulatory role of eNOS on various aspects of cardiac contraction, relaxation and rate has become exceedingly complex since its initial identification in cardiac myocytes. The availability of mouse strains with genetic deletion (or overexpression) of specific NOS (e.g. eNOS) allowed a critical re-examination of previous paradigms, including at the whole organ level in vivo, both in normal and stressed conditions. It appears that endogenous production of NO by eNOS has little influence on the inotropic or lusitropic state of the heart under basal conditions, but that it consistently opposes the inotropic response to β-adrenergic stimulation. The latter is mediated by eNOS activation through β3-adrenergic receptors, which were identified in cardiomyocytes from several species, including man. This countervailing influence of the β3-adrenergic eNOS pathway on the more classical β1-2-adrenergic positive inotropic effect may protect the heart against the toxicity of excessive catecholamine stimulation. The influence of β3-adrenergically stimulated eNOS must be contrasted with that of cardiomyocyte stretch, which also activates eNOS with a resultant increase in EC coupling gain, calcium transient and contraction force that participates in the length-dependent contractile reserve of the heart. These apparently contrasting effects of eNOS may coordinately influence overall cardiac function depending on the specific stimulus that activates the enzyme and the subcellular compartment where it is activated.
The majority of studies examining the role of eNOS in ‘stressed’ (mostly ischaemic) hearts conclude that eNOS is protective, both through its vasodilating and anti-thrombotic actions in the coronary vasculature and its critical triggering of iNOS expression and subsequent essential NO production for late preconditioning. Likewise, eNOS opposes the adverse remodelling after myocardial infarction on the short term, but does not influence mortality on longer term, probably owing to alternative compensatory mechanisms. The challenge is now to translate the substantial body of experimental evidence accumulated over the past 10 years into useful therapies by exploiting the beneficial properties of eNOS-derived NO adapted to specific clinical situations.
References
- Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011–3016. doi: 10.1161/01.cir.0000019516.31040.2d. [DOI] [PubMed] [Google Scholar]
- Balligand JL. Regulation of cardiac beta-adrenergic response by nitric oxide. Cardiovasc Res. 1999;43:607–620. doi: 10.1016/s0008-6363(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Balligand JL. Heat shock protein 90 in endothelial nitric oxide synthase signaling: following the lead(er)? Circ Res. 2002;90:838–841. doi: 10.1161/01.res.0000018173.10175.ff. [DOI] [PubMed] [Google Scholar]
- Balligand JL, Kobzik L, Han X, Kaye DM, Belhassen L, O'Hara DS, Kelly RA, Smith TW, Michel T. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J Biol Chem. 1995;270:14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Bartunek J, Shah AM, Vanderheyden M, Paulus WJ. Dobutamine enhances cardiodepressant effects of receptor-mediated coronary endothelial stimulation. Circulation. 1997;95:90–96. doi: 10.1161/01.cir.95.1.90. [DOI] [PubMed] [Google Scholar]
- Bates TE, Loesch A, Burnstock G, Clark JB. Mitochondrial nitric oxide synthase: a ubiquitous regulator of oxidative phosphorylation? Biochem Biophys Res Commun. 1996;218:40–44. doi: 10.1006/bbrc.1996.0008. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Nat l Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. J Physiol. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RM, Yellon DM. The contribution of endothelial nitric oxide synthase to early ischaemic preconditioning: the lowering of the preconditioning threshold. An investigation in eNOS knockout mice. Cardiovasc Res. 2001;52:274–280. doi: 10.1016/s0008-6363(01)00394-7. [DOI] [PubMed] [Google Scholar]
- Bett GC, Dai S, Campbell DL. Cholinergic modulation of the basal L-type calcium current in ferret right ventricular myocytes. J Physiol. 2002;542:107–117. doi: 10.1113/jphysiol.2002.017335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal DK, Stull JT, Gill GN. Phosphorylation of cardiac troponin by guanosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1978;253:324–326. [PubMed] [Google Scholar]
- Boknik P, Heinroth-Hoffmann I, Kirchhefer U, Knapp J, Linck B, Luss H, Muller T, Schmitz W, Brodde O, Neumann J. Enhanced protein phosphorylation in hypertensive hypertrophy. Cardiovasc Res. 2001;51:717–728. doi: 10.1016/s0008-6363(01)00346-7. [DOI] [PubMed] [Google Scholar]
- Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104:3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choate JK, Danson EJ, Morris JF, Paterson DJ. Peripheral vagal control of heart rate is impaired in neuronal NOS knockout mice. Am J Physiol. 2001;281:H2310–2317. doi: 10.1152/ajpheart.2001.281.6.H2310. [DOI] [PubMed] [Google Scholar]
- Cotton JM, Kearney MT, MacCarthy PA, Grocott-Mason RM, McClean DR, Heymes C, Richardson PJ, Shah AM. Effects of nitric oxide synthase inhibition on basal function and the force-frequency relationship in the normal and failing human heart in vivo. Circulation. 2001;104:2318–2323. doi: 10.1161/hc4401.098515. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem. 2002;277:38079–38086. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- Elvan A, Rubart M, Zipes DP. NO modulates autonomic effects on sinus discharge rate and AV nodal conduction in open-chest dogs. Am J Physiol. 1997;272:H263–271. doi: 10.1152/ajpheart.1997.272.1.H263. [DOI] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- Feron O, Dessy C, Opel DJ, Arstall MA, Kelly RA, Michel T. Modulation of the endothelial nitric-oxide synthase-caveolin interaction in cardiac myocytes. Implications for the autonomic regulation of heart rate. J Biol Chem. 1998b;273:30249–30254. doi: 10.1074/jbc.273.46.30249. [DOI] [PubMed] [Google Scholar]
- Feron O, Michel JB, Sase K, Michel T. Dynamic regulation of endothelial nitric oxide synthase: complementary roles of dual acylation and caveolin interactions. Biochemistry. 1998a;37:193–200. doi: 10.1021/bi972307p. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Flogel U, Decking UK, Godecke A, Schrader J. Contribution of NO to ischemia-reperfusion injury in the saline- perfused heart: a study in endothelial NO synthase knockout mice. JMol Cell Cardiol. 1999;31:827–836. doi: 10.1006/jmcc.1998.0921. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Boissel JP, Kleinert H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III) FASEB J. 1998;12:773–790. [PubMed] [Google Scholar]
- French S, Giulivi C, Balaban RS. Nitric oxide synthase in porcine heart mitochondria: evidence for low physiological activity. Am J Physiol. 2001;280:H2863–2867. doi: 10.1152/ajpheart.2001.280.6.H2863. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- Gauthier C, Leblais, Kobzik L, Trochu JN, Khandoudi N, Bril A, Balligand JL, Le Marec H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102:1377–1384. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- Godecke A, Heinicke T, Kamkin A, Kiseleva I, Strasser RH, Decking UK, Stumpe T, Isenberg G, Schrader J. Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J Physiol. 2001;532:195–204. doi: 10.1111/j.1469-7793.2001.0195g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Kada K, Kamiya K, Toyama J. IGF-I regulates K+-channel expression of cultured neonatal rat ventricular myocytes. Am J Physiol. 1997;272:H2599–2606. doi: 10.1152/ajpheart.1997.272.6.H2599. [DOI] [PubMed] [Google Scholar]
- Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci U S A. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol. 2000;278:H971–981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghada A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh C, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Kobzik L, Balligand JL, Kelly RA, Smith TW. Nitric oxide synthase (NOS3)-mediated cholinergic modulation of Ca2+ current in adult rabbit atrioventricular nodal cells. Circ Res. 1996;78:998–1008. doi: 10.1161/01.res.78.6.998. [DOI] [PubMed] [Google Scholar]
- Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J Gen Physiol. 1995;106:45–65. doi: 10.1085/jgp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan RL, John MC, Kouretas PC, Hack BD, Matherne GP, Laubach VE. Deletion of endothelial nitric oxide synthase exacerbates myocardial stunning in an isolated mouse heart model. J Surg Res. 2000;93:127–132. doi: 10.1006/jsre.2000.5953. [DOI] [PubMed] [Google Scholar]
- Hare JM, Keaney JF, Jr, Balligand JL, Loscalzo J, Smith TW, Colucci WS. Role of nitric oxide in parasympathetic modulation of beta-adrenergic myocardial contractility in normal dogs. Journal of Clinical Investigation. 1995;95:360–366. doi: 10.1172/JCI117664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart CY, Hahn EL, Meyer DM, Burnett JC, Jr, Redfield MM. Differential effects of natriuretic peptides and NO on LV function in heart failure and normal dogs. Am J Physiol. 2001;281:H146–154. doi: 10.1152/ajpheart.2001.281.1.H146. [DOI] [PubMed] [Google Scholar]
- Herring N, Rigg L, Terrar DA, Paterson DJ. NO-cGMP pathway increases the hyperpolarisation-activated current, If, and heart rate during adrenergic stimulation. Cardiovasc Res. 2001;52:446–453. doi: 10.1016/s0008-6363(01)00425-4. [DOI] [PubMed] [Google Scholar]
- Janssens S P, Simouchi A, Quertermous T, Bloch D B, Bloch K D. Cloning and expression of a cDNA encoding human endothelium-derived relating factor/nitric oxide synthase. Journal of Biological Chemistry. 1992;267:14519–14522. published erratum Journal of Biological Chemistry 267, 22694. [PubMed] [Google Scholar]
- Ji GJ, Fleischmann BK, Bloch W, Feelisch M, Andressen C, Addicks K, Hescheler J. Regulation of the L-type Ca2+ channel during cardiomyogenesis: switch from NO to adenylyl cyclase-mediated inhibition. FASEB J. 1999;13:313–324. doi: 10.1096/fasebj.13.2.313. [DOI] [PubMed] [Google Scholar]
- Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, Huang PL, Lefer DJ. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999;276:H1567–1573. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- Jumrussirikul P, Dinerman J, Dawson TM, Dawson VL, Ekelund U, Georgakopoulos D, Schramm LP, Calkins H, Snyder SH, Hare JM, Berger RD. Interaction between neuronal nitric oxide synthase and inhibitory G protein activity in heart rate regulation in conscious mice. J Clin Invest. 1998;102:1279–1285. doi: 10.1172/JCI2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai AJ, Mesaros S, Finkel MS, Oddis CV, Birder LA, Malinski T. β-Adrenergic regulation of constitutive nitric oxide synthase in cardiac myocytes. Am J Physiol. 1997;273:C1371–1377. doi: 10.1152/ajpcell.1997.273.4.C1371. [DOI] [PubMed] [Google Scholar]
- Kanai AJ, Pearce LL, Clemens PR, Birder LA, Vanbibber MM, Choi SY, De Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci U S A. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL, Billiar TR. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation. 2000;101:2742–2748. doi: 10.1161/01.cir.101.23.2742. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Wiviott SD, Kelly RA. Activation of nitric oxide synthase (NOS3) by mechanical activity alters contractile activity in a Ca2+-independent manner in cardiac myocytes: role of troponin I phosphorylation. Biochem Biophys Res Commun. 1999;256:398–403. doi: 10.1006/bbrc.1999.0346. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Noack E. Inhibition of nitric oxide synthase and soluble guanylate cyclase induces cardiodepressive effects in normal rat hearts. Eur J Pharmacol. 1997;334:181–190. doi: 10.1016/s0014-2999(97)01168-0. [DOI] [PubMed] [Google Scholar]
- Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc Res. 1999;42:206–213. doi: 10.1016/s0008-6363(98)00315-0. [DOI] [PubMed] [Google Scholar]
- Kubis N, Besnard S, Silvestre JS, Feletou M, Huang PL, Levy BI, Tedgui A. Decreased arteriolar density in endothelial nitric oxide synthase knockout mice is due to hypertension, not to the constitutive defect in endothelial nitric oxide synthase enzyme. J Hypertens. 2002;20:273–280. doi: 10.1097/00004872-200202000-00017. [DOI] [PubMed] [Google Scholar]
- Kubota I, Han X, Opel DJ, Zhao YY, Baliga R, Huang P, Fishman MC, Shannon RP, Michel T, Kelly RA. Increased susceptibility to development of triggered activity in myocytes from mice with targeted disruption of endothelial nitric oxide synthase. J Mol Cell Cardiol. 2000;32:1239–1248. doi: 10.1006/jmcc.2000.1158. [DOI] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- Lamas S, Marsden PA, Li GK, Tempst P, Michel T. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc Natl Acad Sci U S A. 1992;89:6348–6352. doi: 10.1073/pnas.89.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leesar MA, Stoddard MF, Dawn B, Jasti VG, Masden R, Bolli R. Delayed preconditioning-mimetic action of nitroglycerin in patients undergoing coronary angioplasty. Circulation. 2001;103:2935–2941. doi: 10.1161/01.cir.103.24.2935. [DOI] [PubMed] [Google Scholar]
- Levin KR, Page E. Quantitative studies on plasmalemmal folds and caveolae of rabbit ventricular myocardial cells. Circ Res. 1980;46:244–255. doi: 10.1161/01.res.46.2.244. [DOI] [PubMed] [Google Scholar]
- Levy MN. Sympathetic-parasympathetic interactions in the heart. Circulation Research. 1971;29:437–445. doi: 10.1161/01.res.29.5.437. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Corbin JD. Purified cyclic GMP-dependent protein kinase catalyzes the phosphorylation of cardiac troponin inhibitory subunit (TN-1) J Biol Chem. 1978;253:337–339. [PubMed] [Google Scholar]
- Liu YH, Xu J, Yang JP, Yang F, Shesely E, Carretero OA. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension. 2002;39:375–381. doi: 10.1161/hy02t2.102796. [DOI] [PubMed] [Google Scholar]
- Marletta MA. Nitric oxide synthase structure and mechanism. Journal of Biological Chemistry. 1993;268:12231–12234. [PubMed] [Google Scholar]
- Massion P, Moniotte S, Balligand J. Nitric oxide: does it play a role in the heart of the critically ill? Current Opinion in Critical Care. 2001;7:323–336. doi: 10.1097/00075198-200110000-00003. [DOI] [PubMed] [Google Scholar]
- Matter CM, Mandinov L, Kaufmann PA, Vassalli G, Jiang Z, Hess OM. Effect of NO donors on LV diastolic function in patients with severe pressure-overload hypertrophy. Circulation. 1999;99:2396–2401. doi: 10.1161/01.cir.99.18.2396. [DOI] [PubMed] [Google Scholar]
- Mery PF, Lohmann SM, Walter U, Fischmeister R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mery PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem. 1993;268:26286–26295. [PubMed] [Google Scholar]
- Michel JB, Feron O, Sase K, Prabhakar P, Michel T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem. 1997;272:25907–25912. doi: 10.1074/jbc.272.41.25907. [DOI] [PubMed] [Google Scholar]
- Mohan P, Brutsaert DL, Paulus WJ, Sys SU. Myocardial contractile response to nitric oxide and cGMP. Circulation. 1996;93:1223–1229. doi: 10.1161/01.cir.93.6.1223. [DOI] [PubMed] [Google Scholar]
- Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- Muller-Strahl G, Kottenberg K, Zimmer HG, Noack E, Kojda G. Inhibition of nitric oxide synthase augments the positive inotropic effect of nitric oxide donors in the rat heart. J Physiol. 2000;522:311–320. doi: 10.1111/j.1469-7793.2000.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musialek P, Lei M, Brown HF, Paterson DJ, Casadei B. Nitric oxide can increase heart rate by stimulating the hyperpolarization-activated inward current, If. Circ Res. 1997;81:60–68. doi: 10.1161/01.res.81.1.60. [DOI] [PubMed] [Google Scholar]
- Musialek P, Rigg L, Terrar DA, Paterson DJ, Casadei B. Role of cGMP-inhibited phosphodiesterase and sarcoplasmic calcium in mediating the increase in basal heart rate with nitric oxide donors. J Mol Cell Cardiol. 2000;32:1831–1840. doi: 10.1006/jmcc.2000.1216. [DOI] [PubMed] [Google Scholar]
- Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, Alexander RW, Murphy TJ. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90:2092–2096. doi: 10.1172/JCI116092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla R, Curtis MJ. Effects of NO modulation on cardiac arrhythmias in the rat isolated heart. Circ Res. 1995;77:984–992. doi: 10.1161/01.res.77.5.984. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Paulus WJ. The role of nitric oxide in the failing heart. Heart Failure Reviews. 2001;6:105–118. doi: 10.1023/a:1011453809750. [DOI] [PubMed] [Google Scholar]
- Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89:2070–2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- Paulus WJ, Vantrimpont PJ, Shah AM. Paracrine coronary endothelial control of left ventricular function in humans. Circulation. 1995;92:2119–2126. doi: 10.1161/01.cir.92.8.2119. [DOI] [PubMed] [Google Scholar]
- Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, Sollott SJ. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- Piech A, Dessy C, Havaux X, Feron O, Balligand JL. Differential regulation of nitric oxide synthase and their allosteric regulators in heart and vessels of hypertensive rats. Cardiovasc Res. 2002a doi: 10.1016/s0008-6363(02)00676-4. in the Press. [DOI] [PubMed] [Google Scholar]
- Piech A, Massart PE, Dessy C, Feron O, Havaux X, Morel N, Vanoverschelde JL, Donckier J, Balligand JL. Decreased expression of myocardial eNOS and caveolin in dogs with hypertrophic cardiomyopathy. Am J Physiol. 2002b;282:H219–231. doi: 10.1152/ajpheart.2002.282.1.H219. [DOI] [PubMed] [Google Scholar]
- Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM. Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem. 1992;267:24173–24176. [PubMed] [Google Scholar]
- Rakhit A, Maguire CT, Wakimoto H, Gehrmann J, Li GK, Kelly RA, Michel T, Berul CI. In vivo electrophysiologic studies in endothelial nitric oxide synthase (eNOS)-deficient mice. J Cardiovasc Electrophysiol. 2001;12:1295–1301. doi: 10.1046/j.1540-8167.2001.01295.x. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti M P. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Richter K, Buchner J. Hsp90: chaperoning signal transduction. J Cell Physiol. 2001;188:281–290. doi: 10.1002/jcp.1131. [DOI] [PubMed] [Google Scholar]
- Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. doi: 10.1161/hc3601.094298. [DOI] [PubMed] [Google Scholar]
- Sessa WC, Harrison JK, Barber CM, Zeng D, Durieux ME, D'Angelo DD, Lynch KR, Peach MJ. Molecular cloning and expression of a cDNA encoding endothelial cell nitric oxide synthase. J Biol Chem. 1992;267:15274–15276. [PubMed] [Google Scholar]
- Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG. 8-bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ Res. 1994;74:970–978. doi: 10.1161/01.res.74.5.970. [DOI] [PubMed] [Google Scholar]
- Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in eNOS- deficient mice. Am J Physiol. 2002;282:H2422–2426. doi: 10.1152/ajpheart.00855.2001. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Reviews Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. published erratum Nat Reviews Mol Cell Biol 2, 216. [DOI] [PubMed] [Google Scholar]
- Sumeray MS, Rees DD, Yellon DM. Infarct size and nitric oxide synthase in murine myocardium. J Mol Cell Cardiol. 2000;32:35–42. doi: 10.1006/jmcc.1999.1050. [DOI] [PubMed] [Google Scholar]
- Sunada Y, Ohi H, Hase A, Ohi H, Hosono T, Arata S, Higuchi S, Matsumura K, Shimizu T. Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Human Molecular Genetics. 2001;10:173–178. doi: 10.1093/hmg/10.3.173. [DOI] [PubMed] [Google Scholar]
- Tada H, Thompson CI, Recchia FA, Loke KE, Ochoa M, Smith CJ, Shesely EG, Kaley G, Hintze TH. Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in Langendorff mouse heart. Circ Res. 2000;86:270–274. doi: 10.1161/01.res.86.3.270. [DOI] [PubMed] [Google Scholar]
- Thiemermann C. Corticosteroids and cardioprotection. Nat Med. 2002;8:453–455. doi: 10.1038/nm0502-453. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nat Med. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- Varghese P, Harrison RW, Lofthouse RA, Georgakopoulos D, Berkowitz DE, Hare JM. β3-adrenoceptor deficiency blocks nitric oxide-dependent inhibition of myocardial contractility. J Clin Invest. 2000;106:697–703. doi: 10.1172/JCI9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. 1999;84:1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo Y, Zhang SX, Wu WJ, Wang J, Bao W, Boll R. Ischemic preconditioning upregulates inducible nitric oxide synthase in cardiac myocyte. J Mol Cell Cardiol. 2002;34:5–15. doi: 10.1006/jmcc.2001.1482. [DOI] [PubMed] [Google Scholar]
- Wegener JW, Godecke A, Schrader J, Nawrath H. Effects of nitric oxide donors on cardiac contractility in wild-type and myoglobin-deficient mice. Brit J Pharmacol. 2002;136:415–420. doi: 10.1038/sj.bjp.0704740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmott N, Sethi JK, Walseth TF, Lee HC, White AM, Galione A. Nitric oxide-induced mobilization of intracellular calcium via the cyclic ADP-ribose signaling pathway. J Biol Chem. 1996;271:3699–3705. doi: 10.1074/jbc.271.7.3699. [DOI] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. ProcNatl Acad Sci U S A. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Xuan YT, Tang XL, Qiu Y, Banerjee S, Takano H, Han H, Bolli R. Biphasic response of cardiac NO synthase isoforms to ischemic preconditioning in conscious rabbits. Am J Physiol. 2000;279:H2360–2371. doi: 10.1152/ajpheart.2000.279.5.H2360. [DOI] [PubMed] [Google Scholar]
- Yang XP, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice: cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- Yoo S, Lee SH, Choi BH, Yeom JB, Ho WK, Earm YE. Dual effect of nitric oxide on the hyperpolarization-activated inward current (If) in sino-atrial node cells of the rabbit. J Mol Cell Cardiol. 1998;30:2729–2738. doi: 10.1006/jmcc.1998.0845. [DOI] [PubMed] [Google Scholar]
- Zahradnikova A, Minarovic I, Venema RC, Meszaros LG. Inactivation of the cardiac ryanodine receptor calcium release channel by nitric oxide. Cell Calcium. 1997;22:447–454. doi: 10.1016/s0143-4160(97)90072-5. [DOI] [PubMed] [Google Scholar]
- Ziolo MT, Katoh H, Bers DM. Positive and negative effects of nitric oxide on Ca2+ sparks: influence of β-adrenergic stimulation. Am J Physiol. 2001;281:H2295–2303. doi: 10.1152/ajpheart.2001.281.6.H2295. [DOI] [PubMed] [Google Scholar]
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Investig. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]