

Abstract
Previous studies show that exercise-induced hyperaemia is unaffected by systemic inhibition of nitric oxide synthase (NOS) and it has been proposed that this may be due to compensation by other vasodilators. We studied the involvement of cytochrome P450 2C9 (CYP 2C9) in the regulation of skeletal muscle blood flow in humans and the interaction between CYP 2C9 and NOS. Seven males performed knee extensor exercise. Blood flow was measured by thermodilution and blood samples were drawn frequently from the femoral artery and vein at rest, during exercise and in recovery. The protocol was repeated three times on the same day. The first and the third protocols were controls, and in the second protocol either the CYP 2C9 inhibitor sulfaphenazole alone, or sulfaphenazole in combination with the NOS inhibitor Nω-monomethyl-l-arginine (l-NMMA) were infused. Compared with control there was no difference in blood flow at any time with sulfaphenazole infusion (P > 0.05) whereas with infusion of sulfaphenazole and l-NMMA, blood flow during exercise was 16 ± 4 % lower than in control (9 min: 3.67 ± 0.31 vs. 4.29 ± 0.20 l min−1; P < 0.05). Oxygen uptake during exercise was 12 ± 3 % lower (9 min: 525 ± 46 vs. 594 ± 24 ml min−1; P < 0.05) with co-infusion of sulfaphenazole and l-NMMA, whereas oxygen uptake during sulfaphenazole infusion alone was not different from that of control (P > 0.05). The results demonstrate that CYP 2C9 plays an important role in the regulation of hyperaemia and oxygen uptake during exercise. Since inhibition of neither NOS nor CYP 2C9 alone affect skeletal muscle blood flow, an interaction between CYP 2C9 and NOS appears to exist so that a CYP-dependent vasodilator mechanism takes over when NO production is compromised.
Despite a substantial effort to identify locally formed vasoactive compounds, responsible for the large increases in blood flow to skeletal muscle that occur with exercise, this issue remains largely unsolved. In many tissues, including skeletal muscle, endothelial-derived hyperpolarizing factors (EDHF) have been identified as factors that, independently of NO and prostanoids, can hyperpolarize smooth muscle, and thereby elicit vasodilatation, in response to substances that enhance endothelial calcium levels, e.g. acetylcholine and bradykinin (Bolz et al. 1999; Fisslthaler et al. 2000). Several different EDHFs appear to exist in different tissues and vessels (Quilley et al. 1997), although in coronary arteries of several species (Pinto et al. 1987; Hecker et al. 1994) the EDHF phenomenon involves a cytochrome P450 (CYP) epoxygenase-derived product. In porcine coronary arteries, a CYP 2C isoform homologous to CYP 2C9 plays a crucial role in the generation of the bradykinin-induced, endothelium-dependent hyperpolarization of vascular smooth muscle cells and the subsequent NO synthase- and cyclooxygenase-independent vasodilatation (Fisslthaler et al. 2000).
In human skeletal muscle, bradykinin has been reported to induce hyperaemia independently of NO and prostanoids, suggesting an EDHF-mediated mechanism (Halcox et al. 2000) Recent in vitro studies have, moreover, demonstrated that a product of a CYP 2C enzyme homologous to CYP 2C8 and CYP 2C9 regulates EDHF-mediated responses in hamster skeletal muscle resistance arteries (Bolz et al. 2000). Thus, CYP 2C9 is clearly a potential candidate in the regulation of exercise hyperaemia in humans.
We and others have previously shown that inhibition of NO synthesis by systemic infusion of either Nω-monomethyl-l-arginine (l-NMMA; Shoemaker et al. 1997; Bradley et al. 1999; Rådegran & Saltin, 1999) or Nω-nitro-l-arginine methyl ester (l-NAME; Frandsen et al. 2001) in humans does not alter blood flow during exercise. These observations could indicate redundancy, i.e. that the compromised synthesis of NO can be compensated for by the enhanced formation of other vasodilators. Although the vasodilators adenosine, PGI2 or potassium do not appear to be compounds responsible for a redundancy effect during NOS blockade (Frandsen et al. 2000), other vasoactive substances, such as 11,12-EET (11,12-epoxyeicosatrienoic acid), could be of importance as there is evidence suggesting that NO can inhibit CYP 2C (Bauersachs et al. 1996; Fleming et al. 2001). Thus, the formation of CYP 2C-derived products may be enhanced when NO synthesis is diminished, thereby maintaining blood flow during exercise.
In the present study we examined the role of CYP 2C9 in the regulation of skeletal muscle blood flow at rest and during exercise in humans by arterial infusion of sulfaphenazole. Sulfaphenazole is a highly specific inhibitor of CYP 2C9 (Mancy et al. 1996; Fleming et al. 2001) that has been shown to have no direct effect on K+Ca channel activity in either endothelial or smooth muscle cells (Fisslthaler et al. 1999, 2000; Fleming et al. 2001). To elucidate a possible interaction between CYP 2C9 and NO synthase, arterial infusion of sulfaphenazole was also combined with simultaneous infusion of the NO synthase inhibitor l-NMMA.
Methods
Subjects
Seven healthy male subjects aged between 19 and 29 years, with an average height of 182 cm (range: 176-192 cm), and an average body mass of 76.3 kg (64.1-86.3 kg), participated in the study. The subjects were fully informed of any risks and discomforts associated with the experiments before giving their written informed consent to participate. The experiments were carried out in accordance with the guidelines contained in the Declaration of Helsinki. The study was approved by the Ethics Committee of Copenhagen and Frederiksberg communities.
Experimental design and procedures
The exercise model used was the one-legged knee extensor model that allows the exercise to be confined to the quadriceps muscle (Andersen et al. 1985). Before the experiment the subjects practised the exercise on several occasions.
On the experimental day, two femoral arterial and one femoral venous catheter were placed under local anaesthesia. In the non-experimental leg a femoral arterial catheter was positioned 10-20 mm proximal to the inguinal ligament for blood sampling and measurements of blood pressure. In the experimental leg, a femoral arterial catheter, for the infusion of sulfaphenazole and l-NMMA, and a femoral venous catheter, for blood sampling and for measurements of blood temperature, were placed 1-2 cm distal to the inguinal ligament. The thermistor (Edslab probe 94-0.30-2.5F) was inserted and advanced 80-100 mm proximal to the tip. ECG electrodes were placed on the chest to measure heart rate.
Experimental protocol
The experimental protocol consisted of a 10 min rest period, a 10 min one-legged knee extensor exercise period at work rate of 30 W and a 10 min recovery period, repeated three times on the same day. The work rate of 30 W was selected to be the highest possible that could be performed for the required time by all subjects. During each of the three trials, blood samples were drawn from the femoral artery and vein at 3, 6 and 9 min of rest, after 1, 3, 5, 7 and 9 min of exercise as well as 1, 2, 5 and 9 min into the recovery period. Before and after blood sampling, femoral venous blood flow was measured by the thermodilution technique (Andersen & Saltin, 1985; Gonzáles-Alonso et al. 2000). A cuff was placed below the knee to avoid contribution of blood from the lower leg.
The first part of the protocol was performed without infusion (CON1SULF), the second part with femoral arterial infusion of sulfaphenazole (SULF; Clinalfa, Laufelfingen, Switzerland) and the third part again without infusion (CON2SULF). The first and the second parts were separated by 1 h and the second and third parts were separated by 2 h. The sulfaphenazole was infused at a constant rate of 0.29 mg min−1 (l thigh volume)−1 at rest and at 2.87 mg min−1 (l thigh volume)−1 during the first 6 min of exercise. Infusion was terminated after 6 min in order to examine whether the effect of inhibition on flow was abolished upon cessation of infusion.
On a separate occasion the whole protocol was repeated, but this time, instead of infusion of sulfaphenazole alone, l-NMMA was first infused for 10 min at rest, and sulfaphenazole was then co-infused with l-NMMA for another 10 min at rest and for 6 min during exercise (LN + SULF). l-NMMA was infused at a constant rate of 5 mg min−1 (l thigh volume)−1 for the first 5 min (bolus) and thereafter at a rate of 1 mg min−1 (l thigh volume)−1 for the following 15 min at rest and 6 min of exercise. Sulfaphenazole was infused as in SULF. The data obtained in SULF or in LN + SULF were compared with the mean of the values obtained during CON1 and CON2 of the respective protocol (CONSULF and CONLN+SULF).
Before exercise, the leg was passively moved for 5 s in order to accelerate the flywheel to obtain a constant power output from onset of exercise and the kicking frequency was kept constant at 60 r.p.m. by a metronome. Force tracings from a strain-gauge as well as kicking frequency were continuously recorded during each of the exercise periods.
Blood analysis
Oxygen saturation of blood and haemoglobin concentration was determined spectrophotometrically (Radiometer OSM-3 Hemoximeter). PO2, PCO2 and pH were measured with the Astrup technique (ABL 30, Radiometer, Copenhagen, Denmark). Lactate was analysed on haemolysed blood (Foxdal et al. 1992).
Determination of thigh volume
The volume of the quadriceps femoris muscle group was estimated anthropometrically (Jones & Pearson, 1969) and corrected based on a comparison between MR scan and anthropometric determinations. The mean thigh volume of the experimental leg was 9.29 l (range: 7.70-9.97 l).
Immunohistochemistry
Frozen human skeletal muscle biopsy samples, cut in 8 μm thick sections, were air-dried and fixed in ice-cold acetone for 30 s. Serial sections were stained for CYP 2C9 with primary antibody (458209; Gentest, Woburn, MA, USA) and for endothelial cells with primary antibody CD31 (M0823; DAKO A/S, Glostrup, Denmark). The antibodies were incubated for 1 h and binding was visualized by incubation with a secondary antibody coupled to biotin, followed by a streptavidin-FITC complex.
Statistics
Data are means ± standard error of the mean (s.e.m.). Two-way analysis of variance with repeated measures was used for evaluation of changes during the exercises between CON and SULF and CON and LN + SULF. One-way ANOVA was used to determine the effect of l-NMMA infusion at rest and Newman-Keuls post hoc tests were used to locate differences. A P-value less than 0.05 was regarded as significant.
Results
Thigh blood flow
In CONSULF thigh blood flow at rest was 0.45 ± 0.07 l min−1. It increased during exercise to 3.85 ± 0.44 l min−1 at 9 min and decreased during recovery to 1.50 ± 0.23 l min−1 at 1 min (P < 0.05, Fig. 1A). In SULF thigh blood flows at rest, during exercise and in recovery were similar to those in CONSULF (P > 0.05; Fig. 1A).
Figure 1. Thigh blood flow and vascular conductance in subjects at rest, during knee extensor exercise (30 W) and in recovery.
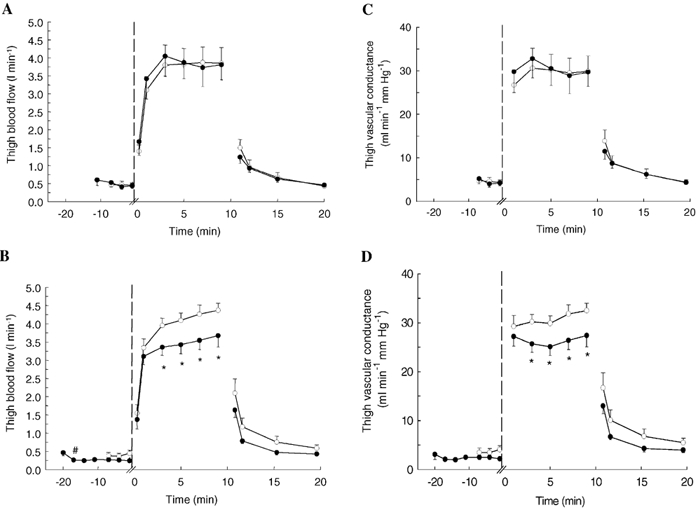
Measurements were performed during control conditions, with infusion of the CYP 2C9 inhibitor sulfaphenazole, and with infusion of sulfaphenazole in combination with the NO synthase inhibitor Nω-monomethyl-l-arginine (l-NMMA). Blood flow during control conditions (○) and infusion of sulfaphenazole (•) (A). Blood flow during control conditions (○) and infusion of l-NMMA and sulfaphenazole (•) (B). Vascular conductance during control conditions (○) and infusion of sulfaphenazole (•) (C). Vascular conductance during control conditions (○) and infusion of l-NMMA and sulfaphenazole (•) (D). #P < 0.05 vs. pre-infusion; *P < 0.05vs. control.
In LN + SULF, infusion of l-NMMA alone at rest lowered thigh blood flow from 0.46 ± 0.08 to 0.28 ± 0.04 l min−1 (P < 0.05), but flow at rest was not further lowered with additional infusion of sulfapenazole (P > 0.05; Fig. 1B). During exercise in LN + SULF, thigh blood flow during 3-9 min of exercise was on average 16± 4 % lower than in CONLN+SULF, with levels reaching 3.67 ± 0.31 and 4.29 ± 0.20 l min−1, respectively, at 9 min (P < 0.05; Fig. 1B). In recovery, no difference between thigh blood flow in CONLN+SULF and LN + SULF was observed (P > 0.05).
Thigh vascular conductance
Vascular conductance was similar in CONSULF and SULF at all times (P > 0.05; Fig. 1C). In LN + SULF, infusion of l-NMMA alone at rest lowered vascular conductance from 3.1 ± 1.0 to 2.5 ± 0.3 ml min−1 mmHg−1 (P < 0.05) and no further reduction was observed with co-infusion of sulfaphenazole (P > 0.05, Fig. 1D). During exercise in LN + SULF, vascular conductance was on average 16 ± 6 % lower than in CONLN+SULF with levels reaching 27.4 ± 2.3 and 32.5 ± 1.5 ml min−1 mmHg−1, respectively, at 9 min (P < 0.05).
Thigh oxygen uptake
In CONSULF, femoral oxygen extraction (a-vdiff,O2) was 75 ± 5 ml l−1 at rest and increased during exercise (5 min: 142 ± 6 ml l−1, P < 0.05; Fig. 2A). The levels of a-vdiff,O2 at rest and during exercise in SULF were similar to those in CONSULF (P > 0.05). In recovery in CONSULF, a-vdiff,O2 had decreased to 73 ± 4 ml l−1 (P < 0.05) after 1 min, and the levels in SULF were not different from those in CONSULF (P > 0.05). In CONSULF, thigh oxygen uptake was 35 ± 7 ml min−1 at rest, increased during exercise (9 min: 545 ± 64 ml min−1, P < 0.05) and decreased again in recovery (P < 0.05; Fig. 2C). The corresponding levels during SULF were not different from those in CONSULF (P > 0.05; Fig. 2C).
Figure 2. Muscle oxygen extraction and oxygen uptake determined from measurements of arterial and venous oxygen content differences and blood flow in subjects at rest, during knee extensor exercise (30 W) and in recovery.
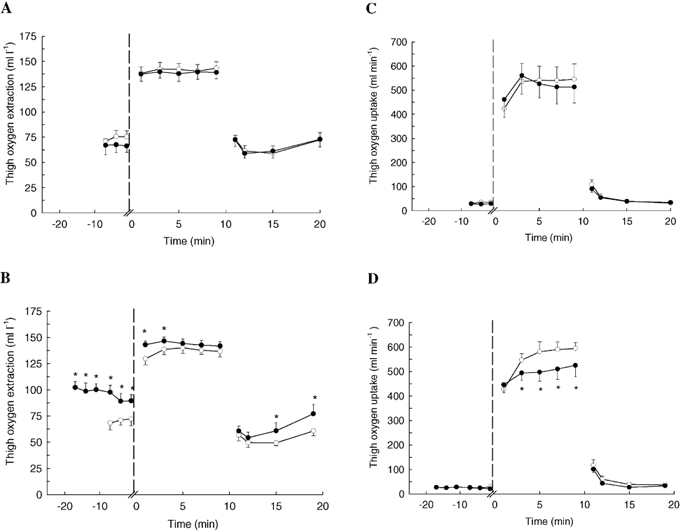
Measurements were performed during control conditions, with infusion of the CYP 2C9 inhibitor sulfaphenazole, and with infusion of sulfaphenazole in combination with the NO synthase inhibitor Nω-monomethyl-l-arginine (l-NMMA). Oxygen extraction during control conditions (○) and infusion of sulfaphenazole (•) (A). Oxygen extraction during control conditions (○) and infusion of l-NMMA and sulfaphenazole (•) (B). Oxygen uptake during control conditions (○) and infusion of sulfaphenazole (•) (C). Oxygen uptake during control conditions (○) and infusion of l-NMMA and sulfaphenazole (•) (D). *P < 0.05vs. control.
In LN + SULF, oxygen extraction was higher than in CONLN+SULF at rest, during the first 3 min of exercise, and from 5 to 10 min of recovery (P < 0.05; Fig 2B). Thigh oxygen uptake in LN + SULF was similar (P > 0.05) to that in CONLN+SULF at rest and in recovery, but on average 12 ± 3 % lower from 3-9 min of exercise (9 min: 525 ± 46 vs. 594 ± 24 ml min−1, P < 0.05; Fig. 2D).
Blood lactate and pH
Venous-arterial lactate difference (v-adiff,lactate) was 0.1 ± 0.0 mmol l−1 at rest in CONSULF and increased (P < 0.05) to 0.8 ± 0.2 mmol l−1 after 3 min of exercise and had decreased again to 0.2 ± 0.1 mmol l−1 after 10 min of recovery (P < 0.05). There was no net lactate release from the thigh in CONSULF at rest, but a significant release was observed during exercise and in recovery (Fig. 3A). The v-adiff,lactate and lactate release were similar (P > 0.05) in SULF and in CONSULF.
Figure 3. Muscle lactate release determined from measurements of arterial and venous lactate differences and blood flow in subjects at rest, during knee extensor exercise (30 W) and in recovery.
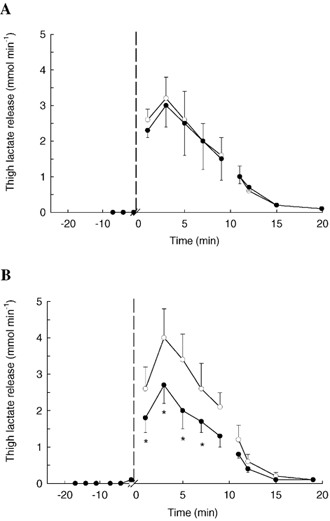
Measurements were performed during control conditions, with infusion of the CYP 2C9 inhibitor sulfaphenazole, and with infusion of sulfaphenazole in combination with the NO synthase inhibitor Nω-monomethyl-l-arginine (l-NMMA). Lactate release during control conditions (○) and infusion of sulfaphenazole (•) (A). Lactate release during control conditions (○) and infusion of l-NMMA and sulfaphenazole (•) (B). *P < 0.05vs. control.
In LN + SULF, v-adiff,lactate at rest was similar (P > 0.05) to in CONLN+SULF but lower from 5-7 min of exercise (5 min: 0.6 ± 0.1 vs. 0.8 ± 0.2 mmol l−1; P < 0.05) and at 2-5 min of recovery (P < 0.05). In LN + SULF, thigh lactate release during exercise was lower than in CONLN+SULF (P < 0.05), whereas no difference was observed at rest or in recovery (Fig. 3B). There were no differences in arterial or venous pH between the trials (P > 0.05).
Heart rate and mean arterial pressure
No differences in heart rate were observed between SULF and CONSULF. In LN + SULF, resting heart rate decreased (P < 0.05) during infusion of l-NMMA alone with no further change with co-infusion of l-NMMA and sulfaphenazole (P > 0.05; Table 1). Prior to exercise, during exercise and in recovery in LN + SULF, heart rate was lower than in CONLN+SULF (P < 0.05). Mean arterial pressures (MAP) at rest, during exercise and in recovery in SULF and LN + SULF were not different from CONSULF and CONLN+SULF, respectively (Table 1).
Table 1.
Mean arterial pressure (MAP)and heart rate (HR), with infusion of sulfaphenazole and with combined infusion of Nω-monomethyl l-arginine (l-NMMA) and sulfaphenazole
| Rest | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Infusion | |||||||||
| No infusion | l-NMMA | Sulfaphenazole | Exercise | Recovery | |||||
| Intervention | 0–5 min | 5–10 min | 5 min | 10 min | 5 min | 10 min | 10 min | 0–2 min | 2–10 min |
| MAP (mmHg) | |||||||||
| Control (CONSULF) | 98 ± 3 | 103 ± 4 | — | — | — | — | 127 ± 4* | 105 ± 4* | 104 ± 4* |
| SULF | — | — | — | — | 104 ± 4 | 106 ± 4 | 125 ± 4 | 109 ± 6 | 104 ± 4 |
| Control (CONLN+SULF) | 106 ± 3 | 107 ± 3 | — | — | — | — | 126 ± 4 | 113 ± 5 | 107 ± 3 |
| LN+SULF | — | — | 105 ± 2 | 108 ± 3 | 105 ± 3 | 107 ± 3 | 127 ± 3 | 112 ± 3 | 109 ± 3 |
| HR (bpm) | |||||||||
| Control (CONLN+SULF) | 56 ± 2 | 58 ± 2 | — | — | — | — | 83 ± 4* | 79 ± 9 | 65 ± 9 |
| SULF | — | — | — | — | 58 ± 3 | 58 ± 2 | 85 ± 4 | 73 ± 4 | 56 ± 2 |
| Control (CON LN+SULF) | 57 ± 1 | 60 ± 1 | — | — | — | — | 89 ± 3 | 80 ± 2 | 62 ± 2 |
| LN+SULF | — | — | 55 ± 2† | 54 ± 2† | 54 ± 2 | 57 ± 2 | 82 ± 3† | 72 ± 3† | 57 ± 3† |
P < 0.05 vs rest
P < 0.05 vs. CONLN+SULF
Distribution of CYP 2C9 in human skeletal muscle
Immunopositive staining for CYP 2C9 was observed in microvascular endothelial cells whereas no staining was detectable in the skeletal muscle cells (Fig. 4).
Figure 4. Distribution of CYP 2C9 in microvascular endothelium (white arrows) of human skeletal muscle.

Inset shows negative control. Scale bar represents 50 μm.
Discussion
The results of the present investigation demonstrate that the simultaneous inhibition of CYP 2C9 and NOS leads to a significant reduction in blood flow and vascular conductance during exercise. This observation indicates that CYP 2C9-derived intermediates play an important role in the regulation of exercise hyperaemia in humans. In a previous study, using the same dose of l-NMMA as in the current study, we have shown that NOS blockade does not alter exercise-induced hyperaemia in humans (Rådegran & Saltin, 1999). Thus, as inhibition of CYP 2C9 alone in the present study also did not affect exercise-induced increases in blood flow, our data further suggest that there is a close interaction between a CYP-derived product and NO formation, and it is likely that this CYP-derived product can compensate for an impaired synthesis of NO to maintain an adequate blood supply to the muscle. Moreover, oxygen extraction during the combined inhibition of CYP 2C9 and NO synthase was not sufficiently elevated to compensate for the reduced blood flow, thus oxygen uptake during exercise was lower than in control.
Many recent studies have concentrated on elucidating the potential role of NO in the regulation of skeletal muscle blood flow during exercise. Although there has been some disagreement with regard to the effect of NO synthase inhibition on exercise-induced hyperaemia, most studies performed in humans, using either thermodilution or the ultrasound Doppler technique for blood flow determination, report no effect of NOS inhibition (Shoemaker et al. 1997; Bradley et al. 1999; Frandsen et al. 2001). The difficulty in identifying crucial vasodilators probably lies in the concept of redundancy, which implies that no effect on flow can be observed when the production of only one vasodilator is inhibited. In the present study co-infusion of the CYP 2C9 inhibitor sulfaphenazole and the NOS inhibitor l-NMMA resulted in a significant decrease in muscle blood flow and vascular conductance during exercise. Neither sulfaphenazole nor l-NMMA (Rådegran & Saltin, 1999) alone altered exercise-induced changes in blood flow. Taken together, these observations highlight two important aspects. The first is the identification of a CYP-dependent mechanism in the regulation of exercise-induced changes in skeletal muscle blood flow; the other is the in vivo documentation of redundancy between NO- and CYP-derived products, most probably EETs. Although the inhibition of NO synthase and CYP 2C9 only partially reduced the exercise-induced blood flow, our observation demonstrates for the first time that blood flow in the exercising human muscle can be altered by this double blockade and that CYP P450 metabolites are likely to be involved in the regulation of flow. This observation opens up the possibility that also other, possibly related, compounds are involved so that when NO synthase and CYP 2C9 are inhibited they compensate and maintain blood flow relatively high, and it will be important to continue to search for such additional compounds.
At rest blood flow and vascular conductance were lowered with l-NMMA infusion alone, to a similar extent as observed in previous studies using NOS blockade (Shoemaker et al. 1997; Rådegran & Saltin, 1999). As l-NMMA alone has no effect on vascular conductance during exercise (Rådegran & Saltin, 1999) the change in conductance occurring with blockade at rest appears to be overcome during exercise. Thus, as co-infusion of l-NMMA and sulfaphenazole did not reduce conductance at rest below that observed with NOS blockade alone, the decrease in conductance with co-infusion during exercise is likely to be independent of that at rest.
EETs, such as 11,12- and 14,15-EET, which are generated by CYP 2C9 (Bylund et al. 1998), have previously been identified as hyperpolarizing factors in the porcine coronary artery (Fisslthaler et al. 2000) as well as several other vascular beds (Quilley et al. 1997; DeWit et al. 1999). However, as neither the membrane potential nor EET production could be determined in the present study it is not possible to conclude that a specific EET, e.g. 11,12-EET, in human skeletal muscle elicits relaxation by inducing the hyperpolarization of vascular smooth muscle cells or via another mechanism. Nevertheless, bradykinin-mediated, nitric oxide- and prostaglandin-independent forearm vasodilatation has been shown to be suppressed by high intravascular K+ concentrations suggesting the involvement of a hyperpolarizing factor in human skeletal muscle vasculature (Halcox et al. 2000). Furthermore, Bolz and co-workers (2000) demonstrated that a CYP 2C isoform, highly homologous to CYP 2C8 and CYP 2C9, appears to be involved in the acetylcholine-induced generation of EDHF responses in hamster gracilis muscle. It therefore appears likely that the CYP-dependent vasodilating effect described in the present study is related to the generation of a hyperpolarizing factor such as EET in human skeletal muscle, and that the vasodilator response is initiated by the hyperpolarization of vascular smooth muscle cells.
The stimulus for the activation of CYP 2C9 in skeletal muscle tissue during exercise is not certain. It has been demonstrated that a cytochrome P450-dependent EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice (Huang et al. 2001), and that cyclic stretch enhances CYP 2C9 activity in endothelial cells (Fisslthaler et al. 2001) and increases the generation of a cytochrome P450-dependent EDHF from the porcine coronary artery (Popp et al. 1998; Paolocci et al. 2001). Thus, considering the observed localization of CYP 2C9 in the vascular endothelium in human skeletal muscle, it appears plausible that pulsatile stretch and fluid shear stress are stimuli for the generation of cytochrome P450 metabolites in human muscle. However, it cannot be excluded that also other, yet unknown, stimuli for CYP 2C9 activation in human muscle tissue may exist.
There was some individual variation in the response to infusion of sulfaphenazole in combination with l-NMMA. In order to examine whether the response in blood flow was dose-dependent the experiment with dual infusion of l-NMMA and sulfaphenazole was repeated in two subjects: one high responder and one low responder. The dose of sulfaphenazole was doubled whereas the l-NMMA dose was kept unaltered. In the low-responder the reduction in blood flow compared with control was 4 % with the lower dose and 23 % with the higher dose of sulfaphenazole, whereas in the high-responder the reduction in flow was similar with the high and with the low dose (11 and 12 %). This finding indicates that whereas the initial dose of sulfaphenazole infused in the present study is adequate for some individuals, others require a greater amount for full inhibition of CYP 2C9.
In the present study we observed that the lower flow during exercise with inhibition of both NOS and CYP 2C9 was not sufficiently compensated for by an increase in oxygen extraction and, thus, muscle oxygen uptake was lower than in control. This was most likely not due to an inability of the muscle to increase extraction as skeletal muscle has a maximal capacity for oxygen extraction of about 180 ml l−1 and extraction in LN + SULF was only about 145 ml l−1. The lowering of oxygen uptake appears to have been the result of the combined inhibition of CYP 2C9 and NO synthase as neither CYP 2C9 inhibition alone nor NO synthase inhibition alone (Frandsen et al. 2001) affect oxygen uptake in humans. In animals and isolated mitochondria, inhibition of NO synthase has been shown to increase oxygen uptake (Shen et al. 1994; Brown, 1995) and the mechanism behind the current finding of reduced oxygen uptake during the combined blockade of CYP 2C9 and NOS must therefore be different from that observed in animals treated with a NOS inhibitor alone. The decrease in oxygen uptake during inhibition of NOS and CYP 2C9 was accompanied by a lower lactate release from the exercising leg compared with control, implying that the anaerobic energy contribution was also lowered. It therefore appears that the efficiency of the muscle was improved, either by an enhanced contractile efficiency or by an elevated P/O ratio in the mitochondria. Although CYP 2C9 was observed to be localized in the capillary endothelium and not in the skeletal muscle cells, it is not unlikely that the CYP 2C9 metabolite could affect the muscle cells. There are several examples of signalling between parenchymal and vascular cells and it has, for example, been shown that eNOS present in the vascular endothelium affects muscle mitochondrial respiration and consequently oxygen uptake (Shen et al. 1995).
Another possibility is that the lowered oxygen uptake was compensated for by a higher anaerobic energy production and that the efficiency of the muscle was unaltered during the double blockade, despite the lowered release of lactate. As muscle biopsies were not obtained in the present study it cannot be excluded that the muscle lactate concentration was increased during the double blockade and that the lactate transport was markedly reduced during this condition. However, as lactate release was unaltered with sulfaphenazole infusion alone and with l-NMMA infusion alone (Rådegran & Saltin, 1999), it seems unlikely that the combination of these inhibitors would have decreased lactate transport. Thus, it appears probable that the mechanical efficiency is elevated when both CYP 2C9 and NO synthase are inhibited simultaneously, but the mechanism behind this effect remains unclear.
During the inhibition with l-NMMA and sulfaphenazole, heart rate was reduced by approximately 8 beats min−1 during exercise, whereas blood pressure remained unaltered, suggesting that the lowered heart rate was not an effect of a baroreceptor reflex. Therefore, as sulfaphenazole or l-NMMA (Rådegran & Saltin, 1999) infusion alone had no effect on exercise blood flow or heart rate, a probable explanation for the lowered heart rate during exercise, with the combined inhibition, is the reduction in blood flow to the leg. Whether the mechanism underlying the lowering of heart rate when blood flow was reduced is related to the alteration in oxygen uptake and/or energy metabolism remains to be elucidated.
In conclusion, the results of the present study demonstrate that a CYP 2C9-dependent product plays an important role in the regulation of skeletal muscle blood flow. The effect of this compound is only revealed upon dual inhibition of NOS and CYP 2C9. Based on this finding and observations from in vitro models in the literature, we propose that there is redundancy between a CYP 2C9-derived product and NO in that the CYP 2C9 metabolite can take over as vasodilator when NO formation is diminished. Thus, in vivo, where certain disease states are known to reduce the bioavailability of NO, CYP 2C9 metabolites, such as 11,12-EET, may compensate for the loss of NO and maintain blood flow to the working muscle.
Acknowledgments
The study was supported by a grant from The Danish National Research Foundation (504-14). We thank Merete Vannby, Ingelise Kring and Winnie Taagerup for excellent technical assistance.
References
- Andersen P, Adams RP, Sjogaard G, Thorboe A, Saltin B. Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol. 1985;59:1647–1653. doi: 10.1152/jappl.1985.59.5.1647. [DOI] [PubMed] [Google Scholar]
- Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol. 1985;366:233–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Bolz S-S, De Wit C, Pohl U. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acethylcholine-induced dilation of microvessels. Br J Pharmacol. 1999;128:124–134. doi: 10.1038/sj.bjp.0702775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz SS, Fisslthaler B, Pieperhoff S, De Wit C, Fleming I, Busse R, Pohl U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB Journal. 2000;14:255–260. doi: 10.1096/fasebj.14.2.255. [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Kingwell BA, McConell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes. 1999;48:1815–1821. doi: 10.2337/diabetes.48.9.1815. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- Bylund J, Ericsson J, Oliw EH. Analysis of cytochrome P450 metabolites of arachidonic and linoleic acids by liquid chromatography-mass spectrometry with ion trap MS. Anal Biochem. 1998;265:55–68. doi: 10.1006/abio.1998.2897. [DOI] [PubMed] [Google Scholar]
- de Wit C, Esser N, Lehr HA, Bolz S, Pohl U. A pentobarbital sensitive EDHF co-mediates Ach-induced arteriolar dilation in the hamster microcirculation. Am J Physiol. 1999;276:H1527–1534. doi: 10.1152/ajpheart.1999.276.5.H1527. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Hinsch N, Chataigneau T, Popp R, Kiss L, Busse R, Fleming I. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor-mediated responses in coronary arteries. Hypertension. 2000;36:270–275. doi: 10.1161/01.hyp.36.2.270. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Michaelis UR, Kiss L, Fleming I, Busse R. Cyclic stretch enhances the expression and activity of the coronary EDHF synthase. Hypertension. 2001;38:1427–1432. doi: 10.1161/hy1201.096532. [DOI] [PubMed] [Google Scholar]
- Fleming I, Michaelis R, Bredenkötter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- Foxdal P, Bergqvist Y, Eckerbom S, Sandhagen B. Improving lactate analysis with the YSI 2300 GL: hemolyzing blood samples makes results comparable with those for deproteinized whole blood. Clin Chem. 1992;38:2110–2114. [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Langberg H, Saltin B, Hellsten Y. Inhibition of nitric oxide synthesis by systemic N(G)-monomethyl-l-arginine administration in humans: effects on interstitial adenosine, prostacyclin and potassium concentrations in resting and contracting skeletal muscle. J Vasc Res. 2000;37:297–302. doi: 10.1159/000025743. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise induced hyperemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with N(G) -nitro-l-arginine methyl ester. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzáles-Alonso J, Quistorff B, Krustrup P, Bangsbo J, Saltin B. Heat production in human skeletal muscle at the onset of intense dynamic exercise. J Physiol. 2000;524:603–615. doi: 10.1111/j.1469-7793.2000.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halcox JPJ, Narayanan S, Cramer-Joyce L, Mincemoyer R, Quyyumi AA. Characterization of endothelium derived hyperpolarizing factor in the human forearm microcirculation. Am J Physiol. 2000;280:H2470–2477. doi: 10.1152/ajpheart.2001.280.6.H2470. [DOI] [PubMed] [Google Scholar]
- Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta A, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol. 2001;280:H2462–2469. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- Jones PR, Pearson J. Anthropometric determination of leg fat and muscle plus bone volumes in young male and female adults. J Physiol. 1969;204.P:63–66P. [PubMed] [Google Scholar]
- Mancy A, Dijols S, Poli S, Guengerich P, Mansuy D. Interaction of sulfaphenazole derivatives with human liver cytochromes P4502C: Molecular origin of the specific inhibitory effects of sulfaphenazole on CYP 2C9 and consequences for the substrate binding site topology of CYP 2C9. Biochemistry. 1996;35:16 205–16 212. doi: 10.1021/bi961950t. [DOI] [PubMed] [Google Scholar]
- Paolocci N, Pagliaro P, Isoda T, Saavedra F W, Kass D A. Role of calcium-sensitive K+ channels and nitric oxide in in vivo coronary vasodilation from enhanced perfusion pulsatility. Circulation. 2001;103:119–124. doi: 10.1161/01.cir.103.1.119. [DOI] [PubMed] [Google Scholar]
- Pinto A, Abraham NG, Mullane KM. Arachidonic acid induced endothelial-dependent relaxations of canine coronary arteries: contribution of a cytochrome P450 dependent pathway. J Pharm Exp Ther. 1987;240:856–882. [PubMed] [Google Scholar]
- Popp R, Fleming I, Busse R. Pulsatile stretch elicits the release of the endothelium-derived hyperpolarizing factor from isolated coronary arteries: a modulator of arterial compliance. Circ Res. 1998;82:696–703. doi: 10.1161/01.res.82.6.696. [DOI] [PubMed] [Google Scholar]
- Quilley J, Fulton D, McGiff JC. Hyperpolarizing factors. Biochem Pharmacol. 1997;54:1059–1070. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- Rådegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol. 1999;276:H1951–1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Shen W, Hintze TH, Wolin MS. An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation. 1995;92:3505–3512. doi: 10.1161/01.cir.92.12.3505. [DOI] [PubMed] [Google Scholar]
- Shen W, Xu X, Ochoa M, Zhao G, Wolin MS, Hintze TH. Role of nitric oxide in the regulation of oxygen consumption in conscious dogs. Circ Res. 1994;75:1086–1095. doi: 10.1161/01.res.75.6.1086. [DOI] [PubMed] [Google Scholar]
- Shoemaker JK, Halliwell JR, Hughson JR, Joyner MJ. Contributions of acethylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol. 1997;273:H2388–2395. doi: 10.1152/ajpheart.1997.273.5.H2388. [DOI] [PubMed] [Google Scholar]