

Abstract
Development of novel therapeutic strategies for congestive heart failure (CHF) seems to be hampered by insufficient knowledge of the molecular machinery of excitation-contraction (EC) coupling in both normal and failing hearts. Cardiac hypertrophy and failure represent a multitude of cardiac phenotypes, and available invasive and non-invasive techniques, briefly reviewed here, allow proper quantification of myocardial function in experimental models even in rats and mice. Both reduced fractional shortening and reduced velocity of contraction characterize myocardial failure. Only when myocardial function is depressed in vivo can meaningful studies be done in vitro of contractility and EC coupling. Also, we point out potential limitations with the whole cell patch clamp technique. Two main factors stand out as explanations for myocardial failure. First, a basic feature of CHF seems to be a reduced Ca2+ load of the sarcoplasmic reticulum (SR) mainly due to a low phosphorylation level of phospholamban. Second, there seems to be a defect of the trigger mechanism of Ca2+ release from the SR. We argue that this defect only becomes manifest in the presence of reduced Ca2+ reuptake capacity of the SR and that it may not be solely attributable to reduced gain of the Ca2+-induced Ca2+ release (CICR). We list several possible explanations for this defect that represent important avenues for future research.
Congestive heart failure (CHF) is an important cause of mortality in Western countries (Ito et al. 2000). It is also a disabling condition. More than 200 years ago Withering systematically described alleviating effects of digitalis on the symptoms of CHF (Withering, 1785). Although digitalis to a large extent has been replaced by other drugs, alternative therapeutic principles are few and even fewer of them target the underlying pathophysiological mechanism directly (Hoshijima & Chien, 2002). Two hallmarks of CHF are cardiac hypertrophy and reduced contractility or function of viable myocardium. Thus the disease seems to directly affect two basic biological processes, control of cardiac growth and the signalling cascade that allows the action potential to trigger contraction (excitation-contraction coupling, or EC coupling). Interestingly, current concepts of these processes in the healthy individual do not comprise enough information to allow us to understand the pathological processes. On the contrary, recent insight into the pathology has in fact provided new understanding of normal biological mechanisms. For example activation of the Ca2+-calmodulin-activated phosphatase calcineurin was first associated with pathological cardiac hypertrophy, but is now recognized as a possible regulator of normal cardiac growth (Wilkins & Molkentin, 2002).
In this review we will therefore focus on the pathology of EC coupling in CHF since in our opinion this literature reveals that our understanding of how the heart beat is normally triggered may be incomplete. We will also point out some methodological restrictions with the patch clamp technique that may have concealed important properties of the EC coupling. More complete reviews of the mechanisms of heart failure have previously been published in entire journal issues (Cardiovascular Research 37(2), 277-548, 1998 and Basic Research in Cardiology 97 (suppl. 1), I/1-I/158, 2002).
Our current concept of the EC coupling in the heart, as recently reviewed by Bers (2002), has developed gradually since Ringer first discovered the dependency of the beating heart on extracellular Ca2+ (1882). Ebashi (1976) and Weber & Murray (1973) initially described the importance of the sarcoplasmic reticulum (SR) in skeletal muscle, and Fabiato (see for instance Fabiato, 1985) introduced the concept of CICR in the heart. Stimulated by the work of Reuter & Seitz (1968), the important relationship between intracellular Na+ and Ca2+ became clear, and it was possible to ascribe the effect of digitalis to inhibition of the Na+-K+ pump which had been discovered by Skou (1957).
The size and duration of the cytosolic Ca2+ transient are important determinants of the velocity, size and duration of cardiomyocyte shortening. The key elements of EC coupling are therefore the recyclable pool of intracellular Ca2+, the velocity of recycling and the mechanism that triggers Ca2+ release from the SR. However, before reviewing these aspects of EC coupling in normal and failing cardiomyocytes, a brief account of the clinical condition is required, since the great disparity of causes of CHF may be reflected in confusing inconsistencies in the literature.
Myocardial function in heart failure
The clinical condition of heart failure
Clinically heart failure is a manifestation of various conditions. A recent definition is that ‘Heart failure is a complex clinical syndrome that can result from any structural or functional cardiac disorder that impairs the ability of the ventricle to fill with or eject blood’ (Hunt et al. 2001). Thus, not all cases of heart failure are due to a reduced myocardial function, which in many cases is reduced contractility of otherwise seemingly healthy myocardium. However, heart failure is a progressive disease, and in many cases irrespective of the cause, myocardial function will eventually deteriorate in the face of unchanged or reduced haemodynamic load (Mann, 1999). It is important that during initial stages the heart failure is compensated and symptoms are scarce (Francis, 2001; Hunt et al. 2001). Also, cardiac performance deteriorates with age (Lakatta, 2002), which means that there could be important interaction between progress of the disease and age related alterations. At some point during the progression of the disease cardiac output can only be maintained at elevated diastolic filling pressure. This leads to pulmonary congestion. Hence, in this review we focus on CHF since it most often means that the disease has progressed to a state where the failure is no longer compensated and the function of seemingly healthy myocardium is depressed. In other words the progression of heart failure can often be linked to an increasing myocardial failure.
Causes of cardiac hypertrophy and heart failure
In principle heart failure has been thought to result from increased wall stress caused either by increased ventricular volume (increased preload) or by increased left ventricular systolic pressure (increased afterload) (Braunwald, 1997). The ensuing hypertrophy will tend to normalize the wall stress, but if hypertrophy and reduced myocardial performance are causally linked, the outcome will still be reduced cardiac performance (Mann, 1999). Such a causal link is difficult to accept since hypertrophy and remodelling of the heart take various shapes. On the cellular level a volume overload will cause serial addition of sarcomeres so the cells mainly become elongated, whereas a pressure overload will cause parallel addition of contractile elements. This means that the growth signals must differ with pressure and volume overload of the left ventricle. Training is a physiological stimulus for cardiac hypertrophy, which will also enhance cardiac performance and has in fact been shown to reduce the cardiac hypertrophy observed in rats after a myocardial infarction (MI) (Wisløff et al. 2002). However, Esposito et al. (2002) recently showed that in genetically modified mice that did not develop hypertrophy in response to pressure overload, cardiac function was better maintained with a high wall stress than in mice that developed hypertrophy. Thus, increased wall stress can be well tolerated for a prolonged time, and their study indicates that at least concentric hypertrophy and reduced contractility may be related. This is in some contrast to an earlier study that showed improved cardiac performance with hypertrophy subsequent to constriction of the transverse aorta (Nakamura et al. 2001). Also Crackower et al. (2002) recently showed that cardiac hypertrophy and contractility defects can be genetically uncoupled. We will not elaborate on the possible link between hypertrophy and contractile properties on the cellular level. This has been reviewed recently (Francis, 2001; Hoshijima & Chien, 2002). We will focus on CHF following MI. In this condition an eccentric hypertrophy develops and an extensive MI with dilatation of the left ventricle may be present, with little sign of congestion and reduced myocardial performance. However, with progression of the disease, CHF develops indicating attenuated contractility of the remaining myocardium (Sjaastad et al. 2000).
Myocardial failure
With echocardiography heart failure can be categorized as systolic and/or diastolic (Hunt et al. 2001). Systolic heart failure is characterized by a reduction in left ventricular performance, reduced contractility, enlargement of the left ventricle, reduction in ejection fraction, and pulmonary congestion. In post-infarction CHF the cardiac systolic function is primarily impaired, but diastolic dysfunction might also be present in the terminally failing post-infarction heart. Patients with primarily diastolic heart failure, such as in hypertensives, typically have pulmonary congestion, but the left ventricular systolic performance can be normal. Even enhanced systolic performance with increased contractility may be present in a diastolic dysfunctional heart. These hearts relax slowly due to low left ventricular compliance, leading to an abnormal left ventricular pressure-volume relationship and elevated left ventricular filling pressure (Zile & Brutsaert, 2002). Thus, the underlying cellular phenotypic changes may be different, underscoring the importance of using well-defined experimental models to unravel mechanisms of altered EC coupling in CHF.
Myocardial systolic dysfunction is characterized by both reduced velocity of contraction and attenuated magnitude of contraction. In vivo this becomes manifest as low ejection fraction and low rate of pressure development (dP/dt) (Carabello, 2002). On the cellular level many investigators only report reduced magnitude of shortening (Wasserstrom et al. 2000; Gomez et al. 2001). This is clearly only one parameter that reflects the in vivo contractility. Figure 1A shows isometric force during twitches of isolated papillary muscles from sham operated (SHAM) and CHF rats. Clearly peak force is reduced in CHF but in addition rate of force development is reduced. This is in agreement with some cellular studies that also report reduced shortening velocities (Ito et al. 2000; Sjaastad et al. 2002b). Thus, in terms of the classical force-velocity concept, power is reduced since both force and velocity are reduced (Fig. 1B) (Alpert et al. 2002). There are reports that fractional shortening (maximum shortening expressed as a percentage of resting cell length) of isolated cells is normal or even increased in CHF (Anand et al. 1997; Prahash et al. 2000) and in hypertrophy with little sign of failure (Litwin & Bridge, 1997; Ito et al. 2000). This of course raises the question of whether such cells are representative of a dysfunctional myocardium. One point to consider is that these cells may still exhibit reduced velocity of shortening, but data on velocity are rarely provided. If velocity of shortening is reduced, power or contractility will also be reduced even in the face of maintained magnitude of shortening.
Figure 1. Cardiomyocyte contractility.
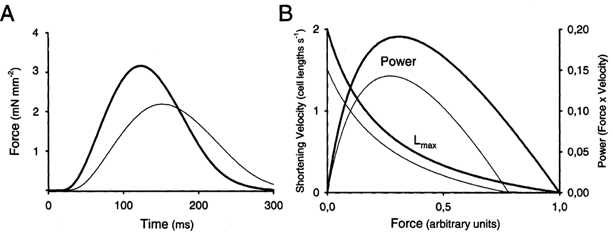
A, contraction relaxation cycles in posterior left ventricular papillary muscles during isometric contractions. Muscles from sham-operated animals (thick line) developed higher force more rapidly than papillary muscles from CHF animals (thin line). B, shortening velocity-force relationship at a muscle length (Lmax) that gives maximal force development. The two examples illustrate contractions in a normal muscle (thick line) and in a muscle with reduced contractility (thin line). The corresponding powers (force × velocity) are illustrated above.
The isometric force and the fractional unloaded shortening are both dependent on the magnitude of the Ca2+ transient. However, the velocity of shortening is closely correlated both to the myofibrillar ATPase activity, which is reduced in heart failure (Alpert et al. 2002), and to the cytosolic Ca2+ concentration (Vannier et al. 1996; Baker et al. 1998; Palmer & Kentish, 1998). Recently it has been pointed out that in heart failure, dyssynchronous release of Ca2+ sparks will lead to a slower rate of rise of the Ca2+ transient (Litwin et al. 2000; Sah et al. 2002), and in a mouse model of hypertrophy and failure both time to peak of the Ca2+ transient and the contraction were observed (Ito et al. 2000). The relative contribution of slowed rate of rise of the Ca2+ transient, reduced peak of the transient and reduced myofibrillar ATPase activity is unclear with regard to causing the reduced velocity of shortening in failing hearts. However, dyssynchronous release of Ca2+ will probably lead to dyssynchronous binding of Ca2+ to troponin C and to slower velocity of contraction. We will therefore focus on possible defects in well-established trigger mechanisms for CICR. We also consider whether a defect in SR function can account for the systolic dysfunction. Finally we discuss the possibility of a defect of other putative trigger mechanisms for SR Ca2+ release. However, some studies on post-infarction myocardial function have utilized animals that did not have CHF, which raises the possibility that these animals did not have myocardial failure (Cheung et al. 1994; Litwin & Bridge, 1997). For this reason we also try to distinguish between post-infarction studies performed on animals with or without a confirmed diagnosis of CHF and we include some comments on methods that have been applied to monitor myocardial function in experimental studies.
Methods for monitoring myocardial function
There is no simple method for evaluating myocardial function in vivo (Carabello, 2002). After an MI the function of the whole heart will not appropriately reflect the contractility of the viable myocardium due to the presence of scar tissue that has variable elasticity. Also experimental models in rat and mice have introduced new challenges with regard to miniaturization of the methods. Since both genetic modification and surgery may lead to great individual variation as to the degree of hypertrophy and failure in experimental models, we stress the importance of individual characterization of myocardial function in vivo before cells or tissue are examined in vitro. Techniques for evaluation of cardiac performance in rats and especially in mice have recently been reviewed so we will only mention a few common procedures (see for instance Christensen et al. 1997; Doevendans et al. 1998; Lorenz, 2002; Janssen et al. 2002; Roth et al. 2002b).
Invasive techniques include measurement of blood pressure, and segment and volume measurements of the heart using piezo-electric crystals or impedance measurements (Esposito et al. 2002). Lately echocardiography has become feasible even in the mouse allowing for monitoring myocardial function. Thus individual animals with various degrees of heart failure can be selected for experiments. Also, with proper consideration of both pre- and afterload, a diagnosis of myocardial failure can be achieved. In the rat MI model left ventricular end diastolic pressure above 15 mmHg correlated very well with reduced shortening velocity of the myocardium, left atrial dilatation and lung congestion (Sjaastad et al. 2000). Even so, direct comparison with the unloaded shortening of isolated cells should be made with great care.
Finally magnetic resonance imaging deserves to be mentioned as a very valuable method, but small bore magnets that provide adequate magnetic fields are still very expensive (Chacko et al. 2000; Weiss, 2001; Wiesmann et al. 2001).
Conclusion
Some of the variability in the literature as regards myocardial and cardiomyocyte function in hypertrophic and failing hearts undoubtedly stems from differences in experimental models. Both invasive and non-invasive techniques allow detailed analysis of cardiac and myocardial function in vivo, which may be valuable for classification of individual animals prior to single cell experiments. Velocity of shortening is equally important for proper function of the heart, as is the degree of shortening. It should be pointed out that myocardial failure (reduced contractility of seemingly intact myocardium) is not necessarily present in the hypertrophied and/or failing heart, especially not after a small myocardial infarction.
Cardiomyocyte contractility in heart failure
In post-infarction rats, shortening of isolated cardiomyocytes has been reported to be both normal (Anand et al. 1997; Prahash et al. 2000) and significantly depressed (Holt et al. 1998; Gomez et al. 2001; Sjaastad et al. 2002b), which could reflect variable experimental models, conditions and techniques. It is therefore difficult to judge on the basis of the literature whether reduced fractional shortening of isolated cells is an inherent property of the failing myocardium. One study showed that with high resistance pipettes and from a holding potential of −70 mV both fractional shortening and velocity of shortening were reduced in CHF cells compared to SHAM cells (Fig. 2A and B) (Sjaastad et al. 2002b). However, when cells were patched with low resistance pipettes using identical protocols there was no longer any difference between CHF and SHAM cells. The reason was that fractional shortening of SHAM cells was lower by 35 % under control conditions with patch pipettes as compared with high resistance pipettes (Fig. 2). Also velocity of shortening was different in patched cells. Thus in this study the patch clamp technique reduced the contractility and velocity of shortening of normal cells, but did not affect contractility of CHF cells. One explanation could be the dialysis of the cells that occurs through low resistance patch pipettes and that the contractile deficit of CHF cells is due to the absence of some substance that is easily dialysed out of SHAM cells, possibly some signalling molecule. With wide bore pipettes the patch clamp technique allows for dialysis of the cell interior, which is a great advantage of the method since it allows for introduction of substances into the cell and maintenance of ion concentrations at desired levels. Hence contractility can easily be manipulated.
Figure 2. Effects of experimental conditions on cell shortening and velocity of shortening.
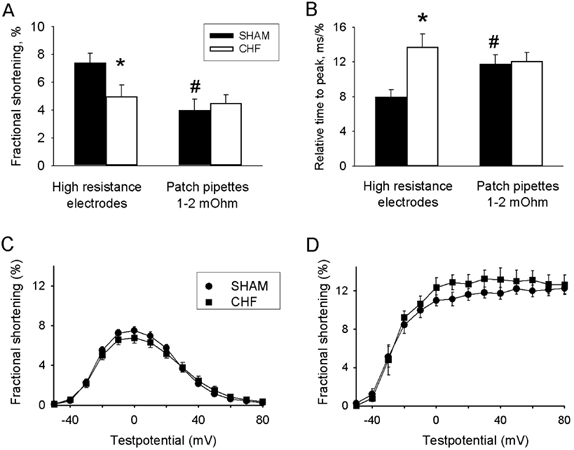
A, fractional shortening and B, velocity of shortening in SHAM cells and CHF cells impaled with high resistance electrodes and low resistance patch pipettes. *Significantly different from SHAM; # significantly different from high resistance electrodes. C, fractional shortening at various voltages using patch pipettes and a post-conditioning potential of −50 mV. Solutions contained Cs+ instead of K+ and there was 0 Na+ in the pipette. D shows the same experiments repeated with K+ containing solutions and 6 mm Na+ in the pipette. From Sjaastad et al. (2002b) with permission.
Other investigators have reported a contractile deficit in cells from rats with myocardial infarction using the same patch clamp procedure (Gomez et al. 2001). This again points either to a true difference in phenotypes because of variable degrees of CHF or duration of the disease, or alternatively to subtle differences in experimental techniques such as different timing of recording after patch rupture, variable degree of dialysis or variable SR Ca2+ load.
Figure 2 also illustrates another aspect of experimental conditions (Sjaastad et al. 2002b). Figure 2C and D shows fractional shortening as a function of test potential using low resistance patch pipettes. With pipette solutions containing a high concentration of Cs+ and no Na+ in the pipette, the shortening-voltage relationship was bell-shaped (Fig. 2C), but when K+ replaced Cs+ and internal Na+ concentration was kept at 6 mm the relationship became sigmoidal (Fig. 2D). This has been ascribed to the effect of reverse mode Na+-Ca2+ exchange at positive potentials (Litwin et al. 1998; Wasserstrom et al. 2000), and contractions at step potentials to positive voltages are attenuated by the drug KBR-793 which inhibits reverse mode Na+-Ca2+ exchange (Piacentino et al. 2000). Importantly, no difference could be observed between CHF and SHAM cells with any of these protocols.
Another important aspect is cellular heterogeneity within the normal and diseased myocardium. The cellular phenotypes are different within the wall of the left ventricle, also in the rat (Gomez et al. 1997a). In addition, after myocardial infarction the wall stress is not homogeneously distributed within the remaining viable myocardium (Loennechen et al. 2001). This latter study also showed that expression of marker genes for hypertrophy and failure was associated with the degree of wall stress. Thus, the region of the left ventricle from which cells are harvested may be very important. Several studies have harvested cells from the border zone between scar tissue in MI hearts and seemingly normal, viable myocardium. These cells show larger hypertrophy and more pronounced deterioration of contractility than cells from a region more remote from the infarct zone (Olivetti et al. 1991; Melillo et al. 1996; Litwin & Bridge, 1997; Kim et al. 2002).
Conclusion
Contractile properties of isolated cardiomyocytes are highly dependent on experimental conditions. It is difficult to reconcile various reports on the contractile deficit of cardiomyocytes from rats with post-infarction CHF. Both reduced and normal fractional shortening have been reported, but few studies report velocities of contraction. There is a possible limitation with the whole cell patch clamp technique, which has been reported to depress contractility of normal cells thus obscuring the contractile deficit of CHF cells. This points to the necessity of using standardized disease models, and carefully monitor and report phenotypes, site within the ventricular wall for cell harvest and experimental conditions to allow comparison of data between studies.
Triggers of calcium release
L-type Ca2+ current (ICa, L)
The inward Ca2+ current through the L-type Ca2+ channels is regarded as the single most important trigger of Ca2+ release (for an extensive treatise see Bers, 2002). Since many studies report proportionality between size of the current, the ensuing Ca2+ transient and contraction (Bers, 2001), reduced current density could in theory be an explanation for reduced contractility. However, as recently reviewed, peak ICa,L density is normal in most types of heart failure (Benitah et al. 2002a) including the post-infarction rat model (Zhang et al. 1995, 1996; Holt et al. 1998; Gomez et al. 2001; Sjaastad et al. 2002b). It has been argued that hypertrophy and failure have opposing effects on the current density (Benitah et al. 2002b) and that the net outcome therefore is unchanged current density. At the single channel level there is a puzzling report that in human heart failure availability and opening probability are both increased as if the channels were hyperphosphorylated (Schröder et al. 1998). However, this could fit with the observation that mRNA and protein expression are reduced and that the size of the gating charge is significantly depressed in a dog model of pacing-induced heart failure which may indicate that the number of channel protein copies is reduced (He et al. 2001). Other observations include slower Ca2+-dependent inactivation and absence of facilitation in human heart failure cells (Barrere-Lemaire et al. 2000). However, in the rat post-infarction model changes in inactivation kinetics could not be observed (Holt et al. 1998). In failing human hearts an isoform switch has been reported for the α1c subunit (Yang et al. 2000), and Chen et al. (2002) recently reported that the density of L-type Ca2+ channels was reduced, but the current maintained due to increased phosphorylation of the channel protein. Clearly, the role of the L-type Ca2+ channel in CHF should be investigated more thoroughly, especially to reconcile a possible discrepancy between single channel and whole cell investigations.
Reverse mode Na+-Ca2+ exchange
Several reports show that reverse mode Na+-Ca2+ exchange can trigger contraction (Nuss & Houser, 1992; Levi et al. 1994; Wasserstrom & Vites, 1996). It has been reported that the exchanger is a less efficient trigger of Ca2+ release than ICa,L (Sipido et al. 1997). Several investigators point out that the exchanger may contribute to increased Ca2+ concentration in the dyadic cleft during the action potential, and hence contribute importantly to stimulate Ca2+ release (Litwin et al. 1998). In a recent simulation study, however, Weber et al. (2002) pointed out that Ca2+ mediated through ICa,L rapidly accumulates in the dyadic cleft, and for that reason the Na+-Ca2+ exchanger runs in reverse mode for a very short time during a normal action potential.
The direction of the Na+-Ca2+ exchange is highly dependent on intracellular Na+ concentration or possibly also a subsarcolemmal pool of Na+ (Lederer et al. 1990). There is evidence that when intracellular Na+ concentration is gradually raised in patch clamp experiments, the exchanger supports contractions at positive test potentials to an increasing extent (Litwin et al. 1998). Therefore the bell-shaped relationship between voltage and contraction that exists when only the L-type Ca2+ channels are providing Ca2+ for triggering release is changed so that contractions are present at positive voltages where L-type Ca2+ current is zero as illustrated in Fig. 2.
In a recent study Lipp et al. (2002) reported that the Ca2+ transients triggered by the L-type Ca2+ current and the Na+ current may not be identical. The L-type Ca2+ current triggered Ca2+ release composed of Ca2+ sparks. A Ca2+ spark is a well-defined quantal release of Ca2+ emanating from a group of Ca2+ release channels (RyRs) (see review by Guatimosim et al. 2002) and can be triggered by a single Ca2+ channel (Santana et al. 1996). However, in the presence of an inward Na+ current Lipp et al. (2002) also detected a Ca2+ release that seemed to be composed of much smaller unitary Ca2+ signals than the sparks and they speculate that this Ca2+ release is triggered by reverse mode Na+-Ca2+ exchange.
Several investigators report that the Na+-Ca2+ exchanger is upregulated by 50-100 % in various models of heart failure and in CHF patients (Studer et al. 1994; Gaughan et al. 1999; Wasserstrom et al. 2000; Barry, 2000) although some investigators have reported unchanged levels in both animal models and patients (Yao et al. 1998; Schwinger et al. 1999b; Piper et al. 2000). An increased expression of the Na+-Ca2+ exchanger can have several important effects: one is reduced Ca2+ load of the SR (see below), another is altered interplay between ICa,L and the exchanger in triggering contraction. Upregulation of the exchanger could make the EC coupling less dependent on ICa,L. In a rat CHF model the effect of verapamil was greatly reduced in CHF cells compared with SHAM at positive potentials above 20 mV (Wasserstrom et al. 2000). In fact verapamil had no significant effect on contractions triggered in this voltage range in CHF cells as opposed to about 60 % reduction of contraction in SHAM. It remains to be seen whether this is of importance when contractions are triggered by action potentials.
Also, upregulation of the Na+-Ca2+ exchanger will make the hearts more prone to Ca2+ overload for instance during ischaemia and reperfusion (Sjaastad et al. 2002a), and it provides a substrate for delayed afterpotentials and lethal arrhythmias (Pogwizd et al. 2001).
Other putative triggers of Ca2+ release
Ca2+ current through T-type Ca2+ channels, TTX-sensitive current through Ca2+ channels, Ca2+ current through Na+ channels and a voltage sensing mechanism have all been proposed as triggers of Ca2+ release (Aggarwal et al. 1997; Santana et al. 1998; Sipido et al. 1998; Zhou & January, 1998; Ferrier & Howlett, 2001). T-type Ca2+ channels are not normally expressed in the left ventricle, but seem to be upregulated in the hypertrophic heart (Martinez et al. 1999). Since they open at more negative voltages than the L-type channels they could contribute to a negative shift of the voltage dependence of contraction. However, this has not been reported in heart failure, and furthermore, these channels would probably contribute to maintain contractions, not reduce them. The TTX-sensitive Ca2+ current is only detectable in the absence of Na+, and the physiological relevance is therefore unclear (Aggarwal et al. 1997). The so-called slip-mode conductance of the Na+ channels allowing Ca2+ to pass only occurs during β-adrenergic stimulation (Santana et al. 1998). However, the existence of this current has been questioned (Piacentino et al. 2002), and there are no studies examining this current in cells from failing hearts. In a series of papers the voltage-sensitive release mechanism (VSRM) has been proposed to be a prominent trigger of contraction (Ferrier et al. 1998; Howlett et al. 1998; Zhu & Ferrier, 2000; Ferrier & Howlett, 2001). It is activated from voltages negative to −40 mV, and contributes to making the contraction-voltage relationship sigmoidal independent of the Na+-Ca2+ exchanger. The VSRM has been reported to be significantly attenuated in cardiomyopathic Syrian hamsters (Howlett et al. 1999). The existence of VSRM is debated (Piacentino et al. 2000), and so far a molecular substrate for such a mechanism has not been identified.
Conclusion
It seems reasonable to conclude that based on existing data the contractile deficit in CHF cannot be ascribed to reduced ICa,L and that in some instances of heart failure upregulation of the Na+-Ca2+ exchanger could in fact reinforce the trigger of contraction. There are conflicting data as to whether contractions triggered by ICa,L are reduced in CHF. It seems that expression of channel protein may be reduced in CHF but that this is compensated for by increased single channel current. Taken together with several reports on various alternative triggers of Ca2+ release than the ICa,L, most notably reverse mode Na+-Ca2+ exchange, it seems important to investigate more closely the role of the L-type Ca2+ channel in the EC coupling.
The intracellular calcium store
The SR Ca2+ store in CHF
For a given peak ICa,L fractional Ca2+ release from the SR increases almost exponentially with SR Ca2+ content (SR load), but no release can be triggered at an SR Ca2+ load of less than 50 % of the maximum load as shown in a study on isolated rabbit cardiomyocytes (Shannon et al. 2000). Therefore, reduced SR Ca2+ load might explain reduced contractility in CHF, primarily reduced fractional shortening. As discussed below, SR load is highly dependent on a number of factors, and it is difficult to distinguish between altered SR load and altered trigger of SR Ca2+ release as an explanation for reduced contractility in CHF. To our knowledge, no studies have really determined the SR Ca2+ content under conditions that prevail in vivo. Studies have been done in isolated cells under variable pacing frequencies by field stimulation or during current or voltage clamp. With a standardized stimulation protocol during voltage clamp in a rat post-infarction CHF model there seems to be no significant difference in SR Ca2+ load by caffeine application (Gomez et al. 2001; Sjaastad et al. 2002b). However, in human heart failure and other experimental models, reduced SR Ca2+ load has been reported (Beuckelmann et al. 1992; Lindner et al. 1998; Pogwizd et al. 2001; Hobai & O'Rourke, 2001). Recently Maier et al. (2002) reported reduced capacity to store Ca2+ in muscle strips from human failing hearts. It is therefore reasonable to assume that in vivo, the SR content of Ca2+ is probably low in CHF contributing to reduced fractional shortening and force development (Pieske et al. 2002).
The Ca2+ pump (SERCA)
Gwathmey et al. (1987) were the first to focus on the reduced capacity of the SERCA to load the SR with Ca2+ in a study of the contractile defect in failing hearts. Later numerous studies examined expression of SERCA at the mRNA and protein levels, as well as the capacity to transport Ca2+. As compiled by Bers (2001, Table 26, p. 319) and Houser et al. (2000) many studies show reduced expression of the SERCA 2 protein, but some also show unaltered expression. Also the results are variable with regard to the regulatory protein phospholamban (Houser et al. 2000; Bers, 2001). We recently showed in one study a nominal decrease of SERCA2 protein and in another study 30 % decrease of SERCA2 protein in the rat post-infarction CHF model (Sande et al. 2002; Sjaastad et al. 2002b). In none of these studies did we find any alteration in phospholamban expression, and the ratio between SERCA2 and phospholamban pentamer or monomer were not significantly changed in CHF.
The expression of SERCA2 and phospholamban in the heart is highly dependent on thyroid hormones. In isolated cardiomyocytes exposed to triiodothyronine for 48 h, phospholamban expression was profoundly reduced and Ca2+ transients were increased (Holt et al. 1999). With more prolonged exposure also SERCA2 level also goes up. The effects of thyroid hormones on the cardiovascular system have recently been reviewed (Klein & Ojamaa, 2001). The point to make here is that not only may thyroid hormones be low in heart failure patients, but also expression of thyroid hormone receptors in the heart is significantly altered (Kinugawa et al. 1999). Thus without careful control of the thyroid status an independent effect of the heart failure condition on expression of SERCA2 and phospholamban will be very difficult to judge. Variable thyroid status may therefore explain some of the variability in the literature regarding expression of these proteins in the failing heart.
Recent focus on SR function has shifted to the phosphorylation status of phospholamban in CHF. There are three phosphorylation sites, of which the serine-16 site and the threonine-17 site are most important. Several studies, both experimental and in humans, have shown a reduced level of serine-16 phosphorylation, suggesting that SERCA2 will be more inhibited than in normal cells (Schmidt et al. 1999; Schwinger et al. 1999a; Sande et al. 2002). This effect seems to predominate over altered expression level, and was also found in non-failing MI rats (Huang et al. 1999). The reduced phosphorylation is associated with significant slowing of Ca2+ reuptake by the SR (Sande et al. 2002). Interestingly in the same study the phosphatases PP1 and PP2A were found to be upregulated, whereas there was no change in the expression of the regulatory unit of protein kinase A. Finally, in line with the reduced level of phosphorylation the response to forskolin was increased in CHF cells. These findings point to an important aberration of intracellular signalling systems in heart failure. Accordingly, overexpression of adenylcyclase in cardiomyopathic mice increases survival (Roth et al. 2002a).
Factors that control the Ca2+ content of SR
SR Ca2+ load is controlled by a multitude of factors that are depicted in Fig. 3. The sarcolemma and the membrane of the sarcoplasmic reticulum are arranged in series so that all ions have to pass both membranes to alter SR Ca2+ load. The concentration of Ca2+ in the cytosol is maintained so low, even though it transiently increases during each beat, that the storage of Ca2+ in the cytosolic compartment is negligible. Altered net transport across the sarcolemma is therefore transmitted to the SR within a few beats. Ca2+ binding proteins and the mitochondria can temporarily store significant amounts of Ca2+, but will not be considered here.
Figure 3. Factors influencing SR Ca2+ load.
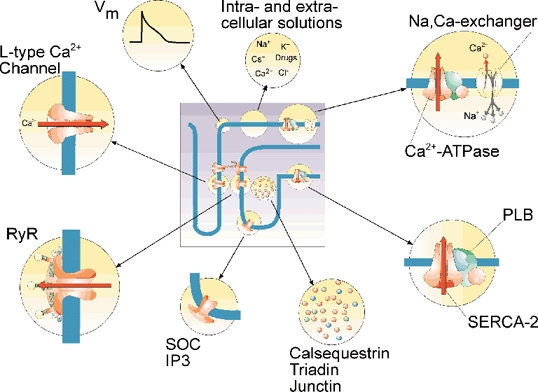
Examples of cellular proteins, cell functions and experimental conditions that influence sarcoplasmic reticulum load are illustrated. SOC, store operated Ca2+ channels; Vm, membrane potential; SERCA-2, sarcoplasmic reticulum Ca2+-ATPase; PLB, phospholamban; RyR, ryanodine receptor. Illustration made by Tore Taraldsen, MD.
The net Ca2+ transfer across the sarcolemma will be controlled by the balance between ICa,L, and reverse and forward modes of Na+-Ca2+ exchange. The outward Ca2+ transport by the sarcolemmal Ca2+-ATPase is very small (Choi & Eisner, 1999). In principle, therefore, it is only temporary mismatches between ICa,L and reverse mode Na+-Ca2+ exchange on one hand and forward mode on the other hand that will cause changes in SR Ca2+ load (Meme et al. 2001). Since cytoplasmic Ca2+ is a common substrate for the Na+-Ca2+ exchanger in forward mode and SERCA2, the two transporters compete for the same cytosolic Ca2+ pool (Bassani et al. 1994; Choi & Eisner, 1999). To put it simply: increased SERCA2 activity will increase SR Ca2+ load and increased Na+-Ca2+ exchange activity will decrease it. Species with high SERCA2 activity will recirculate a large fraction of the Ca2+ inside the cell with each contraction, whereas species with low SERCA2 activity will depend on Ca2+ fluxes across the sarcolemma to provide Ca2+ for the contractile machinery (Bers, 2001).
As cited above, in some studies of the failing and in the hypertrophied heart, independent of species, there is about a doubling of the Na+-Ca2+ exchanger protein (Pogwizd et al. 1999; Wasserstrom et al. 2000; Hobai & O'Rourke, 2000). Thus the slower rate of Ca2+ uptake into the SR (Beuckelmann et al. 1992; Sande et al. 2002) combined with increased expression of exchanger protein will both contribute to reduced SR Ca2+ load. In line with this Shillinger et al. (2000) and Ranu et al. (2002) recently reported that contractions were severely depressed in rabbit cardiomyocytes that overexpress the Na+-Ca2+ exchanger.
However, the picture is complicated by alterations of the driving forces for the Na+-Ca2+ exchanger. Figure 3 indicates that membrane potential and ion gradients play an important role. A depolarisation of 10 mV in right ventricular cardiomyocytes from rats with extensive myocardial infarction as reported by Kaprelian et al. (1999) will significantly shift the balance of the exchanger in favour of more reverse and less forward mode exchange. Furthermore, intracellular Na+ is probably slightly elevated in failing cardiomyocytes (Despa et al. 2002b), which will also favour reverse mode Na+-Ca2+ exchange. Hence, the upregulation of the exchanger protein may well be counterbalanced by reduced driving force for forward Na+-Ca2+ exchange.
Intracellular Na+ concentration is controlled by the Na+-K+ pump. Bundgaard & Keldsen (1996) have reviewed the literature regarding expression levels of the Na+-K+ pump in failing human hearts, and conclude that pump concentration seems to be reduced by about 25 %. One study ascribed this to downregulation of the α1 and β1 subunits as well as a small reduction in α3 (Schwinger et al. 1999b). In an experimental rat study a significant isoform switch was reported in CHF after MI (Semb et al. 1998). Expression of the α2 isoform was reduced and the α3 isoform appeared, whereas the expression of α1 was unchanged. This did not seem to affect the k0.5, which was much higher than in other species. Altered level of pump protein expression relative to tissue weight or tissue protein does not necessarily reflect Na+-K+ pump efficacy in the tissue (Ellingsen et al. 1994). As recently pointed out by Gomez et al. (2002) with respect to the Na+-Ca2+ exchanger and by Semb et al. (1998) as regards the Na+-K+ pump, it is important to consider cell volume and surface to volume ratio when judging the capacity of these transporters to maintain the cytosolic concentrations of Ca2+ and Na+, respectively. In the latter study it was reported that the number of pump protein copies per cell was unchanged. However, since CHF cells were about 30 % larger than SHAM they contained more Na+ and the Na+-K+ pump could not adjust the concentration as rapidly as in SHAM cells. In the study by Ellingsen et al. (1994) it was pointed out that even though the tissue concentration of Na+-K+ pumps was reduced, the number of pumps in the whole left ventricle was unchanged. Despa et al. (2002a) did not find reduced capacity of the Na+-K+ pump in a rabbit heart failure model, but ascribed the higher cytosolic Na+ concentration to an increased leak. It seems safe to conclude that alterations of the Na+-K+ pump expression are not very pronounced in heart failure, but intracellular Na+ seems to be increased by a combined effect of reduced expression of pump protein in some cases, larger cell volume, and increased leak of Na+. Together these effects are large enough to significantly alter the driving force for the Na+-Ca2+ exchanger.
Another important determinant of SR Ca2+ load is the RyR. As neatly pointed out in several papers from Eisner's group (Eisner et al. 2000; Trafford et al. 2000, 2001), a sudden increase or a sudden decrease of release through the RyR for a given stimulus will, within a few beats, cause adjustments of the SR Ca2+ load so that release is again normalized. Thus, provided SERCA2 activity was maintained, a reduced release or gain of the EC coupling in CHF might cause increased SR Ca2+ load so that the size of Ca2+ transients would be maintained. This is clearly not the case, so that by this reasoning it seems that any change in EC coupling would not be sufficient to reduce fractional shortening in CHF unless it is accompanied by reduced capacity for Ca2+ reuptake by the SR. Thus, overexpression of SERCA may restore fractional shortening even in the face of continued inefficiency of the EC coupling (del Monte et al. 1999; Miyamoto et al. 2000; Davia et al. 2001). Synchronization of sparks is probably not related to SR Ca2+ load, and reduced velocity of shortening might therefore be a more sensitive indicator of defective EC coupling than the variable fractional shortening.
Conclusion
SR Ca2+ load is an important determinant of the size of the Ca2+ transient and cardiac contractility. Regulation of SR Ca2+ load is extremely complex, but it seems that in many instances of heart failure, and possibly also CHF, SR Ca2+ load is reduced. However, depolarisation and increased intracellular Na+ concentration might counteract upregulation of the Na+-Ca2+ exchanger and depressed phosphorylation of phospholamban and thus contribute to maintain an adequate Ca2+ content of the SR. Interestingly, a defect in the EC coupling might not be sufficient in itself to cause reduced Ca2+ transients, but requires concomitant reduction of Ca2+ reuptake capacity by the SR.
A variable gain?
Gain of the EC coupling in CHF
As alluded to above, efficiency of the trigger (the size of ICa,L) to cause Ca2+ release from the SR has been termed ‘gain’ of the EC coupling (Beuckelmann & Wier, 1988; Wier et al. 1994; Wier & Balke, 1999). Reduced gain in heart failure has been reported by several authors, as recently reviewed by Benitah et al. (2002b). Gain is dependent not only on the SR Ca2+ load, but also on membrane potential. At negative voltages, although ICa,L is reduced, the driving force for inward Ca2+ current is increased, which seems to make it more efficient in triggering release (Santana et al. 1996).
In many cases of heart failure, reduced gain of the EC coupling seems to be due to reduced SR Ca2+ load (Hobai & O'Rourke, 2001; Pogwizd et al. 2001). However, also under experimental conditions that are aimed at keeping the SR Ca2+ load constant, gain has been reported to be reduced in CHF (Gomez et al. 1997b; Gomez et al. 2001). In another study Sjaastad et al. (2002b) were not able to see this defect. At various temperatures there were no differences in ICa,L between CHF and SHAM rats, and contractions were not significantly different at any voltages between −40 and +60 mV. Also, the contraction- current relationship was almost linear from −40 to +60 mV when contractions were triggered by ICa,L, suggesting that ICa,L-triggered gain was not very high at negative voltages (Sjaastad et al. 2002b). However, possible differences between these studies (Gomez et al. 1997b, 2001; Sjaastad et al. 2002b) include duration of disease, severity of failure and possibly also SR Ca2+ load. Accordingly, some important considerations should be pointed out. First, SR Ca2+ load should be monitored under the exact same conditions that were used for the voltage clamp protocols. Second, contractions and Ca2+ release can be triggered at voltages below −40 mV where the macroscopic ICa,L is very small (Piacentino et al. 2002), or even in the absence of any ICa,L provided a VSRM mechanism exists (Ferrier & Howlett, 2001). Thus, gain may approach infinity and will be very difficult to quantify unless experimental conditions are carefully designed.
Possible causes of altered gain in CHF
The major players in the coupling between the sarcolemma and the SR are depicted in Fig. 4. They comprise first of all the L-type Ca2+ channels (no. 1), and the Na+-Ca2+ exchanger (no. 2) and possibly a VSRM (no. 3).
Figure 4. Summary of alterations in EC coupling in CHF.
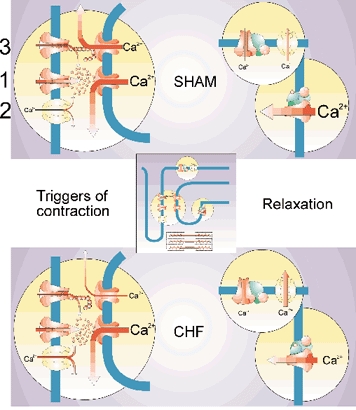
An L-type Ca2+ channel (1), both t-tubular (2) and sarcolemmal Na+-Ca2+ exchangers, a sarcolemmal voltage sensor (3), the sarcoplasmic reticulum Ca2+-ATPase, phospholamban and a sarcolemmal Ca2+-ATPase are illustrated. The following structures have altered function in CHF compared to sham: the Na+-Ca2+ exchanger both in trigger and relaxation mode, the voltage sensor, phospholamban and the sarcoplasmic reticulum Ca2+-ATPase. Illustration made by Tore Taraldsen, MD.
Since the Na+-Ca2+ exchanger is upregulated it is not likely that reduced gain can be ascribed to altered function of this protein. However, there could be several reasons why L-type Ca2+ channels and RyRs do not communicate efficiently. First it has been reported that the structure of the t-tubules is altered in heart failure (He et al. 2001). Second, a reduced number of ryanodine receptors relative to L-type Ca2+ channels has also been observed (Milnes & MacLeod, 2001). Third, increased distance between the two proteins has been suggested (Gomez et al. 2001). Fourth, increased phosphorylation of the ryanodine receptor has been claimed to alter EC coupling, and cause a basal leak of Ca2+ from the SR (Marx et al. 2000; Marks, 2000; Ono et al. 2000; Yano et al. 2000; Reiken et al. 2001; Doi et al. 2002; Marks et al. 2002). Similarly, the L-type Ca2+ channels may be hyperphosphorylated (Schröder et al. 1998; Chen et al. 2002). This finding might imply very different signalling systems in CHF cells as compared with SHAM since it has now been shown that phosphorylation of phospholamban is reduced and not increased (Schwinger et al. 1999a; Sande et al. 2002). It should also be mentioned that the VSRM has been reported to be highly sensitive to the phosphorylation status of the cell, and seems to be attenuated with reduced phosphorylation levels (Hobai et al. 1997; Ferrier et al. 1998; Zhu & Ferrier, 2000). Finally, there is recently a growing interest in the role of the cytoskeleton in targeting both channel proteins and signalling systems that control EC coupling (Zile et al. 2001; Rueckschloss & Isenberg, 2001). This may be a new avenue for understanding the defects of EC coupling in heart failure.
Conclusion
Gain of the EC coupling in heart failure is clearly reduced by low SR Ca2+ content, but also, independently of load, some investigators have reported reduced gain of the EC coupling although this is not a universal finding. Both the L-type Ca2+ channel and the RyR seem to be hyperphosphorylated in CHF. Several explanations, some involving altered structure of t-tubules and/or the dyad, have been proposed. Very few studies have addressed the question of whether reduced velocity of shortening in CHF can be linked to altered gain or efficiency of the EC coupling. Clearly our current concepts of the molecular machinery that constitutes the link between the t-tubules and the SR need to be refined.
Perspectives
Cardiac hypertrophy and failure represent a multitude of phenotypes, and future studies should carefully monitor myocardial function in vivo to allow interpretation of data from in vitro experiments. Even with well-defined reduced contractility of the myocardium reflected on the cellular level, there is as yet no single explanation for the contractile deficit. Rather, an improved understanding of EC coupling calls for further investigations to unravel both the normal and the pathological trigger mechanisms of cardiomyocyte contraction.
References
- Aggarwal R, Shorofsky SR, Goldman L, Balke CW. Tetrodotoxin-blockable calcium currents in rat ventricular myocytes; a third type of cardiac cell sodium current. J Physiol. 1997;505:353–369. doi: 10.1111/j.1469-7793.1997.353bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert NR, Mulieri LA, Warshaw D. The failing human heart. Cardiovasc Res. 2002;54:1–10. doi: 10.1016/s0008-6363(02)00248-1. [DOI] [PubMed] [Google Scholar]
- Anand IS, Liu D, Chugh SS, Prahash AJ, Gupta S, John R, Popescu F, Chandrashekhar Y. Isolated myocyte contractile function is normal in postinfarct remodeled rat heart with systolic dysfunction. Circulation. 1997;96:3974–3984. doi: 10.1161/01.cir.96.11.3974. [DOI] [PubMed] [Google Scholar]
- Baker AJ, Figueredo VM, Keung EC, Camacho SA. Ca2+ regulates the kinetics of tension development in intact cardiac muscle. Am J Physiol. 1998;44:H744–750. doi: 10.1152/ajpheart.1998.275.3.H744. [DOI] [PubMed] [Google Scholar]
- Barrere-Lemaire S, Piot C, Leclercq F, Nargeot J, Richard S. Facilitation of L-type calcium currents by diastolic depolarization in cardiac cells: impairment in heart failure. Cardiovasc Res. 2000;47:336–349. doi: 10.1016/s0008-6363(00)00107-3. [DOI] [PubMed] [Google Scholar]
- Barry WH. Na+-Ca2+ exchange in failing myocardium: friend or foe? Circ Res. 2000;87:529–531. doi: 10.1161/01.res.87.7.529. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah JP, Gomez AM, Fauconnier J, Kerfant BG, Perrier E, Vassort G, Richard S. Voltage-gated Ca2+ currents in the human pathophysiologic heart: a review. Basic Res Cardiol. 2002a;97(suppl. 1):I/11–I/18. doi: 10.1007/s003950200023. [DOI] [PubMed] [Google Scholar]
- Benitah JP, Kerfant BG, Vassort G, Richard S, Gomez AM. Altered communication between L-type calcium channels and ryanodine receptors in heart failure. Front Biosci. 2002b;7:e263–e275. doi: 10.2741/benitah. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001. [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Wier WG. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J Physiol. 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunwald E. Heart Disease. A Textbook of Cardiovascular Medicine. 5. Philadelphia: W.B. Saunders Company; 1997. [Google Scholar]
- Bundgaard H, Kjeldsen K. Human myocardial Na,K-ATPase concentration in heart failure. Mol Cell Biochem. 1996;163–164:277–283. doi: 10.1007/BF00408668. [DOI] [PubMed] [Google Scholar]
- Carabello BA. Evolution of the study of left ventricular function: everything old is new again. Circulation. 2002;105:2701–2703. doi: 10.1161/01.cir.0000021240.86593.9d. [DOI] [PubMed] [Google Scholar]
- Chacko VP, Aresta F, Chacko SM, Weiss RG. MRI/MRS assessment of in vivo murine cardiac metabolism, morphology, and function at physiological heart rates. Am J Physiol. 2000;279:H2218–2224. doi: 10.1152/ajpheart.2000.279.5.H2218. [DOI] [PubMed] [Google Scholar]
- Chen X, Piacentino V, III, Furukawa S, Goldman B, Margulies KB, Houser SR. L-Type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–524. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- Cheung JY, Musch TI, Misawa H, Semanchick A, Elensky M, Yelamarty RV, Moore RL. Impaired cardiac function in rats with healed myocardial infarction: cellular vs. myocardial mechanisms. Am J Physiol. 1994;266:C29–36. doi: 10.1152/ajpcell.1994.266.1.C29. [DOI] [PubMed] [Google Scholar]
- Choi HS, Eisner DA. The effects of inhibition of the sarcolemmal Ca-ATPase on systolic calcium fluxes and intracellular calcium concentration in rat ventricular myocytes. Pflugers Arch. 1999;437:966–971. doi: 10.1007/s004240050868. [DOI] [PubMed] [Google Scholar]
- Christensen G, Wang Y, Chien KR. Physiological assessment of complex cardiac phenotypes in genetically engineered mice. Am J Physiol. 1997;272:H2513–2524. doi: 10.1152/ajpheart.1997.272.6.H2513. [DOI] [PubMed] [Google Scholar]
- Crackower M, Oudit G, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng H, Rybin V, Lembo G, Fratta L, Oliveira-dos-Santos A, Benovic J, Kahn C, Izumo S, Steinberg S, Wymann M, Backx P, Penninger J. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Davia K, Bernobich E, Ranu HK, del Monte F, Terracciano CM, Macleod KT, Adamson DL, Chaudhri B, Hajjar RJ, Harding SE. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2001;33:1005–1015. doi: 10.1006/jmcc.2001.1368. [DOI] [PubMed] [Google Scholar]
- del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002a;539:133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002b;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Doevendans PA, Daemen MJ, De Muinck ED, Smits JF. Cardiovascular phenotyping in mice. Cardiovasc Res. 1998;39:34–49. doi: 10.1016/s0008-6363(98)00073-x. [DOI] [PubMed] [Google Scholar]
- Doi M, Yano M, Kobayashi S, Kohno M, Tokuhisa T, Okuda S, Suetsugu M, Hisamatsu Y, Ohkusa T, Kohno M, Matsuzaki M. Propranolol prevents the development of heart failure by restoring FKBP12. 6-mediated stabilization of ryanodine receptor. Circulation. 2002;105:1374–1379. doi: 10.1161/hc1102.105270. [DOI] [PubMed] [Google Scholar]
- Ebashi S. Excitation-contraction coupling. Annu Rev Physiol. 1976;38:293–313. doi: 10.1146/annurev.ph.38.030176.001453. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Diaz ME, O'Neill SC, Trafford AW. The ryanodine receptor: cause or consequence of diabetic heart failure? J Mol Cell Cardiol. 2000;32:1377–1379. doi: 10.1006/jmcc.2000.1163. [DOI] [PubMed] [Google Scholar]
- Ellingsen Ø, Holthe MR, Svindland A, Aksnes G, Sejersted OM, Ilebekk A. Na,K-pump concentration in hypertrophied human hearts. Eur Heart J. 1994;15:1184–1190. doi: 10.1093/oxfordjournals.eurheartj.a060651. [DOI] [PubMed] [Google Scholar]
- Esposito A, Rapacciuolo A, Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier GR, Howlett SE. Cardiac excitation-contraction coupling: role of membrane potential in regulation of contraction. Am J Physiol. 2001;280:H1928–1944. doi: 10.1152/ajpheart.2001.280.5.H1928. [DOI] [PubMed] [Google Scholar]
- Ferrier GR, Zhu J, Redondo IM, Howlett SE. Role of cAMP-dependent protein kinase A in activation of a voltage-sensitive release mechanism for cardiac contraction in guinea-pig myocytes. J Physiol. 1998;513:185–201. doi: 10.1111/j.1469-7793.1998.185by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GS. Pathophysiology of chronic heart failure. Am J Med. 2001;110(suppl. 7A):37S–46S. doi: 10.1016/s0002-9343(98)00385-4. [DOI] [PubMed] [Google Scholar]
- Gaughan JP, Furukawa S, Jeevanandam V, Hefner CA, Kubo H, Margulies KB, Mcgowan BS, Mattiello JA, Dipla K, Piacentino V, III, Li S, Houser SR. Sodium/calcium exchange contributes to contraction and relaxation in failed human ventricular myocytes. Am J Physiol. 1999;277:H714–724. doi: 10.1152/ajpheart.1999.277.2.H714. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Benitah JP, Henzel D, Vinet A, Lorente P, Delgado C. Modulation of electrical heterogeneity by compensated hypertrophy in rat left ventricle. Am J Physiol. 1997a;272:H1078–1086. doi: 10.1152/ajpheart.1997.272.3.H1078. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Schwaller B, Porzig H, Vassort G, Niggli E, Egger M. Increased exchange current but normal Ca2+ transport via Na+-Ca2+ exchange during cardiac hypertrophy after myocardial infarction. Circ Res. 2002;91:323–330. doi: 10.1161/01.res.0000031384.55006.db. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997b;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- Guatimosim S, Dilly K, Santana LF, Jafri MS, Sobie EA, Lederer WJ. Local Ca2+ signaling and EC coupling in heart: Ca2+ sparks and the regulation of the [Ca2+]i transient. J Mol Cell Cardiol. 2002;34:941–950. doi: 10.1006/jmcc.2002.2032. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Copelas L, Mackinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- Hobai IA, Howarth FC, Pabbathi VK, Dalton GR, Hancox JC, Zhu JQ, Howlett SE, Ferrier GR, Levi AJ. "Voltage-activated Ca release" in rabbit, rat and guinea-pig cardiac myocytes, and modulation by internal cAMP. Pflugers Arch. 1997;435:164–173. doi: 10.1007/s004240050496. [DOI] [PubMed] [Google Scholar]
- Hobai IA, O'Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- Holt E, Sjaastad I, Lunde PK, Christensen G, Sejersted OM. Thyroid hormone control of contraction and the Ca2+-ATPase/phospholamban complex in adult rat ventricular myocytes. J Mol Cell Cardiol. 1999;31:645–656. doi: 10.1006/jmcc.1998.0900. [DOI] [PubMed] [Google Scholar]
- Holt E, Tonnessen T, Lunde PK, Semb SO, Wasserstrom JA, Sejersted OM, Christensen G. Mechanisms of cardiomyocyte dysfunction in heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1581–1593. doi: 10.1006/jmcc.1998.0724. [DOI] [PubMed] [Google Scholar]
- Hoshijima M, Chien KR. Mixed signals in heart failure: cancer rules. J Clin Invest. 2002;109:849–855. doi: 10.1172/JCI15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Xiong W, Mapplebeck CL, Ferrier GR. Role of voltage-sensitive release mechanism in depression of cardiac contraction in myopathic hamsters. Am J Physiol. 1999;277:H1690–1700. doi: 10.1152/ajpheart.1999.277.5.H1690. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Zhu JQ, Ferrier GR. Contribution of a voltage-sensitive calcium release mechanism to contraction in cardiac ventricular myocytes. Am J Physiol. 1998;274:H155–170. doi: 10.1152/ajpheart.1998.274.1.H155. [DOI] [PubMed] [Google Scholar]
- Huang B, Wang S, Qin D, Boutjdir M, El Sherif N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of β-adrenergic pathway, Gi protein, phosphodiesterase, and phosphatases. Circ Res. 1999;85:848–855. doi: 10.1161/01.res.85.9.848. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Baker DW, Chin MH, Cinquegrani MP, Feldmanmd AM, Francis GS, Ganiats TG, Goldstein S, Gregoratos G, Jessup ML, Noble RJ, Packer M, Silver MA, Stevenson LW, Gibbons RJ, Antman EM, Alpert JS, Faxon DP, Fuster V, Gregoratos G, Jacobs AK, Hiratzka LF, Russell RO, Smith SC., Jr ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (committee to revise the 1995 guidelines for the evaluation and management of heart failure). Developed in collaboration with the International Society for Heart and Lung Transplantation; endorsed by the Heart Failure Society of America. Circulation. 2001;104:2996–3007. doi: 10.1161/hc4901.102568. [DOI] [PubMed] [Google Scholar]
- Ito K, Yan X, Tajima M, Su Z, Barry WH, Lorell BH. Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ Res. 2000;87:588–595. doi: 10.1161/01.res.87.7.588. [DOI] [PubMed] [Google Scholar]
- Janssen B, Debets J, Leenders P, Smits J. Chronic measurement of cardiac output in conscious mice. Am J Physiol. 2002;282:R928–935. doi: 10.1152/ajpregu.00406.2001. [DOI] [PubMed] [Google Scholar]
- Kaprielian R, Wickenden AD, Kassiri Z, Parker TG, Liu PP, Backx PH. Relationship between K+ channel down-regulation and [Ca2+]i in rat ventricular myocytes following myocardial infarction. J Physiol. 1999;517:229–245. doi: 10.1111/j.1469-7793.1999.0229z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim SJ, Kramer CM, Yatani A, Takagi G, Mankad S, Szigeti GP, Singh D, Bishop SP, Shannon RP, Vatner DE, Vatner SF. Altered excitation-contraction coupling in myocytes from remodeled myocardium after chronic myocardial infarction. J Mol Cell Cardiol. 2002;34:63–73. doi: 10.1006/jmcc.2001.1490. [DOI] [PubMed] [Google Scholar]
- Kinugawa S, Tsutsui H, Satoh S, Takahashi M, Ide T, Igarashisaito K, Arimura K, Egashira K, Takeshita A. Role of Ca2+ availability to myofilaments and their sensitivity to Ca2+ in myocyte contractile dysfunction in heart failure. Cardiovasc Res. 1999;44:398–406. doi: 10.1016/s0008-6363(99)00205-9. [DOI] [PubMed] [Google Scholar]
- Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev. 2002;7:29–49. doi: 10.1023/a:1013797722156. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- Levi AJ, Spitzer KW, Kohmoto O, Bridge JH. Depolarization-induced Ca entry via Na-Ca exchange triggers SR release in guinea pig cardiac myocytes. Am J Physiol. 1994;266:H1422–1433. doi: 10.1152/ajpheart.1994.266.4.H1422. [DOI] [PubMed] [Google Scholar]
- Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743–749. doi: 10.1006/jmcc.1997.0626. [DOI] [PubMed] [Google Scholar]
- Lipp P, Egger M, Niggli E. Spatial characteristics of sarcoplasmic reticulum Ca2+ release events triggered by L-type Ca2+ current and Na+ current in guinea-pig cardiac myocytes. J Physiol. 2002;542:383–393. doi: 10.1113/jphysiol.2001.013382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin SE, Bridge JH. Enhanced Na+-Ca2+ exchange in the infarcted heart. Implications for excitation-contraction coupling. Circ Res. 1997;81:1083–1093. doi: 10.1161/01.res.81.6.1083. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Li J, Bridge JHB. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys J. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- Loennechen JP, Støylen A, Beisvag V, Wisløff U, Ellingsen Ø. Regional expression of endothelin-1, ANP, IGF-1, and LV wall stress in the infarcted rat heart. Am J Physiol. 2001;280:H2902–2910. doi: 10.1152/ajpheart.2001.280.6.H2902. [DOI] [PubMed] [Google Scholar]
- Lorenz JN. A practical guide to evaluating cardiovascular, renal, and pulmonary function in mice. Am J Physiol. 2002;282:R1565–1582. doi: 10.1152/ajpregu.00759.2001. [DOI] [PubMed] [Google Scholar]
- Maier LS, Braunhalter J, Horn W, Weichert S, Pieske B. The role of SR Ca2+-content in blunted inotropic responsiveness of failing human myocardium. J Mol Cell Cardiol. 2002;34:455–467. doi: 10.1006/jmcc.2002.1527. [DOI] [PubMed] [Google Scholar]
- Mann DL. Mechanisms and models in heart failure: A combinatorial approach. Circulation. 1999;100:999–1008. doi: 10.1161/01.cir.100.9.999. [DOI] [PubMed] [Google Scholar]
- Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- Marks AR, Reiken S, Marx SO. Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation. 2002;105:272–275. [PubMed] [Google Scholar]
- Martinez ML, Heredia MP, Delgado C. Expression of T-type Ca2+ channels in ventricular cells from hypertrophied rat hearts. J Mol Cell Cardiol. 1999;31:1617–1625. doi: 10.1006/jmcc.1999.0998. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Melillo G, Lima JA, Judd RM, Goldschmidt-Clermont PJ, Silverman HS. Intrinsic myocyte dysfunction and tyrosine kinase pathway activation underlie the impaired wall thickening of adjacent regions during postinfarct left ventricular remodeling. Circulation. 1996;93:1447–1458. doi: 10.1161/01.cir.93.7.1447. [DOI] [PubMed] [Google Scholar]
- Meme W, O'Neill SC, Eisner DA. Low sodium inotropy is accompanied by diastolic Ca2+ gain and systolic loss in isolated guinea-pig ventricular myocytes. J Physiol. 2001;530:487–495. doi: 10.1111/j.1469-7793.2001.0487k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, MacLeod KT. Reduced ryanodine receptor to dihydropyridine receptor ratio may underlie slowed contraction in a rabbit model of left ventricular cardiac hypertrophy. J Mol Cell Cardiol. 2001;33:473–485. doi: 10.1006/jmcc.2000.1320. [DOI] [PubMed] [Google Scholar]
- Miyamoto MI, del Monte F, Schmidt U, Disalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97:793–798. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Rokosh DG, Paccanaro M, Yee RR, Simpson PC, Grossman W, Foster E. LV systolic performance improves with development of hypertrophy after transverse aortic constriction in mice. Am J Physiol. 2001;281:H1104–1112. doi: 10.1152/ajpheart.2001.281.3.H1104. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Houser SR. Sodium-calcium exchange-mediated contractions in feline ventricular myocytes. Am J Physiol. 1992;263:H1161–1169. doi: 10.1152/ajpheart.1992.263.4.H1161. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Capasso JM, Meggs LG, Sonnenblick EH, Anversa P. Cellular basis of chronic ventricular remodeling after myocardial infarction in rats. Circ Res. 1991;68:856–869. doi: 10.1161/01.res.68.3.856. [DOI] [PubMed] [Google Scholar]
- Ono K, Yano M, Ohkusa T, Kohno M, Hisaoka T, Tanigawa T, Kobayashi S, Kohno M, Matsuzaki M. Altered interaction of FKBP12. 6 with ryanodine receptor as a cause of abnormal Ca2+ release in heart failure. Cardiovasc Res. 2000;48:323–331. doi: 10.1016/s0008-6363(00)00191-7. [DOI] [PubMed] [Google Scholar]
- Palmer S, Kentish JC. Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and relaxation in rat and guinea pig skinned trabeculae. Circ Res. 1998;83:179–186. doi: 10.1161/01.res.83.2.179. [DOI] [PubMed] [Google Scholar]
- Piacentino V, III, Dipla K, Gaughan JP, Houser SR. Voltage-dependent Ca2+ release from the SR of feline ventricular myocytes is explained by Ca2+-induced Ca2+ release. J Physiol. 2000;523:533–548. doi: 10.1111/j.1469-7793.2000.t01-1-00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentino V, III, Gaughan JP, Houser SR. L-type Ca2+ currents overlapping threshold Na+ currents: could they be responsible for the “slip-mode” phenomenon in cardiac myocytes? Circ Res. 2002;90:435–442. doi: 10.1161/hh0402.105666. [DOI] [PubMed] [Google Scholar]
- Piper C, Bilger J, Henrichs EM, Wudel E, Schultheiss HP, Horstkotte D, Dorner A. Is Na+Ca2+ exchanger expression altered in the endomyocardium of patients with chronic heart valve diseases parallel to myocardial dysfunction? Z Kardiol. 2000;89:682–690. doi: 10.1007/s003920070196. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Prahash AJ, Gupta S, Anand IS. Myocyte response to β-adrenergic stimulation is preserved in the noninfarcted myocardium of globally dysfunctional rat hearts after myocardial infarction. Circulation. 2000;102:1840–1846. doi: 10.1161/01.cir.102.15.1840. [DOI] [PubMed] [Google Scholar]
- Ranu HK, Terracciano CM, Davia K, Bernobich E, Chaudhri B, Robinson SE, Bin KZ, Hajjar RJ, Macleod KT, Harding SE. Effects of Na+/Ca2+-exchanger overexpression on excitation-contraction coupling in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2002;34:389–400. doi: 10.1006/jmcc.2001.1521. [DOI] [PubMed] [Google Scholar]
- Reiken S, Gaburjakova M, Gaburjakova J, He KL, Prieto A, Becker E, Yi GH, Wang J, Burkhoff D, Marks AR. β-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation. 2001;104:2843–2848. doi: 10.1161/hc4701.099578. [DOI] [PubMed] [Google Scholar]
- Reuter H, Seitz N. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J Physiol. 1968;195:451–470. doi: 10.1113/jphysiol.1968.sp008467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer S. A further contribution regarding the influence of different constituents of the blood on the contraction of the heart. J Physiol. 1882;IV:29–42. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002a;105:1989–1994. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- Roth DM, Swaney JS, Dalton ND, Gilpin EA, Ross J., Jr Impact of anesthesia on cardiac function during echocardiography in mice. Am J Physiol. 2002b;282:H2134–2140. doi: 10.1152/ajpheart.00845.2001. [DOI] [PubMed] [Google Scholar]
- Rueckschloss U, Isenberg G. Cytochalasin D reduces Ca2+ currents via cofilin-activated depolymerization of F-actin in guinea-pig cardiomyocytes. J Physiol. 2001;537:363–370. doi: 10.1111/j.1469-7793.2001.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002;90:165–173. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- Sande JB, Sjaastad I, Hoen IB, Bokenes J, Tonnessen T, Holt E, Lunde PK, Christensen G. Reduced level of serine16 phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res. 2002;53:382–391. doi: 10.1016/s0008-6363(01)00489-8. [DOI] [PubMed] [Google Scholar]
- Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78:166–171. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- Santana LF, Gomez AM, Lederer WJ. Ca2+ flux through promiscuous cardiac Na+ channels: slip-mode conductance. Science. 1998;279:1027–1033. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- Schillinger W, Janssen PM, Emami S, Henderson SA, Ross RS, Teucher N, Zeitz O, Philipson KD, Prestle J, Hasenfuss G. Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na+-Ca2+ exchanger. Circ Res. 2000;87:581–587. doi: 10.1161/01.res.87.7.581. [DOI] [PubMed] [Google Scholar]
- Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Human heart failure: cAMP stimulation of SR Ca2+-ATPase activity and phosphorylation level of phospholamban. Am J Physiol. 1999;277:H474–480. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- Schröder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976. doi: 10.1161/01.cir.98.10.969. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca2+-sensitivity of SERCA 2a in failing human myocardium due to reduced serin16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999a;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Wang J, Frank K, Mller-Ehmsen J, Brixius K, McDonough AA, Erdmann E. Reduced sodium pump α1, α3, and β1-isoform protein levels and Na+, K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999b;99:2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- Semb SO, Lunde PK, Holt E, Tønnessen T, Christensen G, Sejersted OM. Reduced myocardial Na+,K+-pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1311–1328. doi: 10.1006/jmcc.1998.0696. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys J. 2000;78:334–343. doi: 10.1016/S0006-3495(00)76596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Carmeliet E, Van De Werf F. T-type Ca2+ current as a trigger for Ca2+ release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. J Physiol. 1998;508:439–451. doi: 10.1111/j.1469-7793.1998.439bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Maes M, Van De Werf F. Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circ Res. 1997;81:1034–1044. doi: 10.1161/01.res.81.6.1034. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Bentzen JG, Semb SO, Ilebekk A, Sejersted OM. Reduced calcium tolerance in rat cardiomyocytes after myocardial infarction. Acta Physiol Scand. 2002a;175:261–269. doi: 10.1046/j.1365-201X.2002.00999.x. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Bøkenes J, Swift F, Wasserstrom JA, Sejersted OM. Normal contractions triggered by ICa,L in ventricular myocytes from rats with post infarction CHF. Am J Physiol. 2002b;283:H1225–1236. doi: 10.1152/ajpheart.00162.2001. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Sejersted OM, Ilebekk A, Bjørnerheim R. Echocardiographic criteria for detection of post-infarction congestive heart failure in rats. J Appl Physiol. 2000;89:1445–1454. doi: 10.1152/jappl.2000.89.4.1445. [DOI] [PubMed] [Google Scholar]
- Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta. 1957;23:394–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Diaz ME, Eisner DA. Coordinated control of cell Ca2+ loading and triggered release from the sarcoplasmic reticulum underlies the rapid inotropic response to increased L-type Ca2+ current. Circ Res. 2001;88:195–201. doi: 10.1161/01.res.88.2.195. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522:259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier C, Chevassus H, Vassort G. Ca-dependence of isometric force kinetics in single skinned ventricular cardiomyocytes from rats. Cardiovasc Res. 1996;32:580–586. [PubMed] [Google Scholar]
- Wasserstrom JA, Holt E, Sjaastad I, Lunde PK, Ødegaard A, Sejersted OM. Altered E-C coupling in rat ventricular myocytes from failing hearts 6 wk after MI. Am J Physiol. 2000;279:H798–807. doi: 10.1152/ajpheart.2000.279.2.H798. [DOI] [PubMed] [Google Scholar]
- Wasserstrom JA, Vites AM. The role of Na+-Ca2+ exchange in activation of excitation-contraction coupling in rat ventricular myocytes. J Physiol. 1996;493:529–542. doi: 10.1113/jphysiol.1996.sp021401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Murray JM. Molecular control mechanisms in muscle contraction. Physiol Rev. 1973;53:612–673. doi: 10.1152/physrev.1973.53.3.612. [DOI] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, III, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- Weiss RG. Imaging the murine cardiovascular system with magnetic resonance. Circ Res. 2001;88:550–551. doi: 10.1161/01.res.88.6.550. [DOI] [PubMed] [Google Scholar]
- Wier WG, Balke CW. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ Res. 1999;85:770–776. doi: 10.1161/01.res.85.9.770. [DOI] [PubMed] [Google Scholar]
- Wier WG, Egan TM, Lopez-lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann F, Ruff J, Engelhardt S, Hein L, Dienesch C, Leupold A, Illinger R, Frydrychowicz A, Hiller KH, Rommel E, Haase A, Lohse MJ, Neubauer S. Dobutamine-stress magnetic resonance microimaging in mice: acute changes of cardiac geometry and function in normal and failing murine hearts. Circ Res. 2001;88:563–569. doi: 10.1161/01.res.88.6.563. [DOI] [PubMed] [Google Scholar]
- Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy: Where have we been? Where are we going? J Physiol. 2002;541:1–8. doi: 10.1113/jphysiol.2002.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withering W. An Account of the Foxglove, and Some of its Medicinal Uses: With Practical Remarks on Dropsy and Other Diseases. London: G.G.J. & J. Robinson; 1785. [Google Scholar]
- Yang Y, Chen X, Margulies K, Jeevanandam V, Pollack P, Bailey BA, Houser SR. L-type Ca2+ channel α1c subunit isoform switching in failing human ventricular myocardium. J Mol Cell Cardiol. 2000;32:973–984. doi: 10.1006/jmcc.2000.1138. [DOI] [PubMed] [Google Scholar]
- Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12. 6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, Muelheims G, Ross JJ, Barry WH. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am J Physiol. 1998;275:H1441–1448. doi: 10.1152/ajpheart.1998.275.4.H1441. [DOI] [PubMed] [Google Scholar]
- Zhang XQ, Moore RL, Tillotson DL, Cheung JY. Calcium currents in postinfarction rat cardiac myocytes. Am J Physiol. 1995;269:C1464–1473. doi: 10.1152/ajpcell.1995.269.6.C1464. [DOI] [PubMed] [Google Scholar]
- Zhang XQ, Tillotson DL, Moore RL, Zelis R, Cheung JY. Na+-Ca2+ exchange currents and SR Ca2+ contents in postinfarction myocytes. Am J Physiol. 1996;271:C1800–1807. doi: 10.1152/ajpcell.1996.271.6.C1800. [DOI] [PubMed] [Google Scholar]
- Zhou Z, January CT. Both T- and L-type Ca2+ channels can contribute to excitation-contraction coupling in cardiac Purkinje cells. Biophys J. 1998;74:1830–1839. doi: 10.1016/S0006-3495(98)77893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Ferrier GR. Regulation of a voltage-sensitive release mechanism by Ca2+-calmodulin-dependent kinase in cardiac myocytes. Am J Physiol. 2000;279:H2104–2115. doi: 10.1152/ajpheart.2000.279.5.H2104. [DOI] [PubMed] [Google Scholar]
- Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Causal mechanisms and treatment. Circulation. 2002;105:1503–1508. doi: 10.1161/hc1202.105290. Part II. [DOI] [PubMed] [Google Scholar]
- Zile MR, Green GR, Schuyler GT, Aurigemma GP, Miller DC, Cooper G. Cardiocyte cytoskeleton in patients with left ventricular pressure overload hypertrophy. J Am Coll Cardiol. 2001;37:1080–1084. doi: 10.1016/s0735-1097(00)01207-9. [DOI] [PubMed] [Google Scholar]