

Abstract
In the guinea pig gastric antrum, the effects of sodium nitroprusside (SNP), an NO donor, on pacemaker potentials were investigated in the presence of nifedipine. The pacemaker potentials consisted of primary and plateau components; SNP (> 1 μm) increased the frequency of occurrence of these pacemaker potentials, while inhibiting the plateau component. 1H-[1,2,4]-Oxadiazole [4,3-a] quinoxalin-1-one, an inhibitor of guanylate cyclase, had no effect on the excitatory actions of SNP on the frequency of pacemaker potentials. Other types of NO donor, (±)-S-nitroso-N-acetylpenicillamine, 3-morpholino-sydnonimine and 8-bromoguanosine 3′5’-cyclic monophosphate had no excitatory effect on pacemaker activity. Forskolin, an activator of adenylate cyclase, or 4,4′-diisothiocyano-stilbene-2,2′-disulphonic acid, an inhibitor of the Ca2+-activated Cl− channel, strongly attenuated the generation of pacemaker potentials, and SNP added in the presence of these chemicals restored the generation of pacemaker potentials. The pacemaker potentials evoked by SNP were abolished in low-Ca2+ solution or by membrane depolarization with high-K+ solution. The SNP-induced generation of pacemaker potentials was not prevented by cyclopiazonic acid, an inhibitor of internal Ca2+-ATPase, but was limited to a transient burst by iodoacetic acid, an inhibitor of glycolysis, carbonyl cyanide m-chlorophenyl-hydrazone, a mitochondrial protonophore, or 1,2-bis(2-aminophenoxy)ethane-N,N,N‘,N‘-tetraacetic acid acetoxymethyl ester, an intracellular Ca2+ chelator. These results suggest that the SNP-induced increase in the frequency of pacemaker potentials is related to the elevated intracellular Ca2+ concentrations due to release from mitochondria, and these actions may be independent of the activation of guanylate cyclase.
Gastric smooth muscles are spontaneously active, and they generate rhythmic activities such as slow waves or action potentials, or both (Tomita, 1981). Thuneberg (1982) considered that the rhythmic activities might originate in the interstitial cells of Cajal (ICC), which are distributed in the myenteric region of gastrointestinal tracts, since these cells are rich in mitochondria and have close contact with surrounding ICC and smooth muscle cells. ICC express c-Kit immunoreactivity and form gap junctional connections with each other and with nearby smooth muscle cells (Komuro et al. 1996; Huizinga et al. 1997; Sanders et al. 1999). ICC are heterogenous, and many types of ICC with different immunohistochemical and electrical properties, such as myenteric ICC (ICC-MY), intramuscular ICC (ICC-IM), deep muscular plexus ICC (ICC-DMP) and submucosal ICC (ICC-SM), are distributed in the gastrointestinal tract (Sanders et al. 1999). In the gastric antrum of the guinea pig, ICC-MY generate periodic plateau-form pacemaker potentials (driving potentials, Dickens et al. 1999). In animal models lacking ICC-MY, rhythmic activity in the small intestine is strongly attenuated, indicating that these cells are indeed essential for pacing gastrointestinal activity (Maeda et al. 1992; Ward et al. 1994; Huizinga et al. 1995). In the guinea pig antrum, simultaneous recordings of electrical responses from ICC-MY and smooth muscle cells confirm that the activity appears first in the former and then propagates rather passively to the latter (Dickens et al. 1999; Hirst & Edwards, 2001).
Nitric oxide (NO) may be the mediator of non-adrenergic, non-cholinergic (NANC) inhibitory nerves in the gastrointestinal smooth muscle, since the inhibitory junction potential evoked by transmural nerve stimulation is inhibited by Nω-nitro-l-arginine, an NO synthase inhibitor, or 1H-[1,2,4]-oxadiazole[4,3-a]quinoxalin-1-one (ODQ), a soluble guanylate cyclase inhibitor (Sanders & Ward, 1992; Kuriyama et al. 1998). In various types of smooth muscles, NO supplied by NO donors such as sodium nitroprusside (SNP) or 3-morpholino-sydnonimine (SIN-1) induces a relaxation, possibly by increasing the production of cyclic guanosine 3′,5′-monophosphate (cGMP) through activation of soluble guanylate cyclase (Lincoln & Cornwell, 1993; Carvajal et al. 2000). However, the actions of these NO donors on gastrointestinal smooth muscles are equivocal, and the excitatory actions of SNP have been noted in some gastrointestinal smooth muscles (Hirano et al. 1997; Holzer et al. 1997; Lefebvre & Bartho, 1997; Alcon et al. 2001; Zhang & Paterson, 2001). In the smooth muscle of the opossum oesophagus and guinea pig gall bladder, SNP induces contraction by activation of excitation- contraction coupling mechanisms (Alcon et al. 2001; Zhang & Paterson, 2001). Thus, the responses of gastrointestinal smooth muscles to NO donors are not identical to those evoked by excitation of NANC inhibitory nerves.
In the present study, the actions of SNP on spontaneous activity in isolated smooth muscles from the guinea pig gastric antrum were investigated. SNP is commonly used as an NO donor, and knowledge of the actions of this chemical on gastric muscle may facilitate understanding of the mechanisms of modulation of gastric motility by NANC inhibitory nerves. The primary aim of these experiments were to investigate the effects of SNP on pacemaker potentials recorded using intracellular microelectrodes. The results indicate that SNP increases the frequency of pacemaker potentials in a cGMP-independent manner. The experiments were further developed to investigate the effects of various types of chemicals such as cyclopiazonic acid (CPA), 1,2-(bis(2-aminophenoxy)ethane-N,N,N‘,N‘-tetraacetic acid acetoxymethyl ester (BAPTA-AM), carbonyl cyanide m-chlorophenyl-hydrazone (CCCP) and iodoacetic acid (IAA), which reportedly modulate intracellular Ca2+ handling mechanisms, on SNP-induced excitatory actions to pacemaker potentials. The results suggest that intracellular Ca2+ and mitochondrial functions are involved in the excitatory actions of SNP on pacemaker potentials.
Methods
Albino guinea pigs of either gender, weighing 200-300 g, were anaesthetized with fluoromethyl 2,2,2-trifluoro-1-(trifluoromethyl) ethyl ether (Sevoflurane, Maruishi Pharmaceuticals, Osaka, Japan) and exsanguinated from the femoral artery. All animals were treated ethically according to the guiding principles for the care and use of animals in the field of physiological sciences, approved by The Physiological Society of Japan. The stomach was excised and opened by cutting along the small curvature in Krebs solution (see below). The mucosal layers were removed by cutting with fine scissors, and the smooth muscle tissues were isolated from the antrum region. The serosal layer was carefully removed with the aid of a dissecting microscope. A tissue segment (about 1.5 mm wide and 3 mm long), with longitudinal and circular muscles attached, was pinned out with the serosal side uppermost on a silicone rubber plate fixed at the bottom of an organ bath (8 mm wide, 8 mm deep, 20 mm long). The tissue was superfused with warmed (35 °C) and oxygenated Krebs solution, at a constant flow rate of about 2 ml min−1. Experiments were carried out in the presence of 1 μm nifedipine throughout, so as to minimize muscle movements.
Conventional microelectrode techniques were used to record intracellular electrical responses of smooth muscle tissues, and the glass capillary microelectrodes (outer diameter 1.2 mm, inner diameter 0.6 mm; Hilgenberg, Germany) filled with 3 m KCl had tip resistances ranging between 50 and 80 MΩ. Electrical responses recorded via a high-input impedance amplifier (Axoclamp-2B, Axon Instruments, Foster City, CA, USA) were displayed on a cathode-ray oscilloscope (SS-7602, Iwatsu, Osaka, Japan) and stored on a personal computer for later analysis.
The ionic composition of the Krebs solution was as follows (mm): Na+ 137.4, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, HCO3− 15.5, H2PO4− 1.2, Cl− 134 and glucose 11.5. Solutions containing high levels of K+ (high-K+ solutions) were prepared by replacing NaCl with KCl. A low-Ca2+ solution ([Ca2+]o = 0.25 mm) was prepared by reducing the volume of CaCl2 to one-tenth. The solutions were aerated with O2 containing 5 % CO2, and the pH of the solutions was maintained at 7.2-7.3.
Drugs used were CCCP, CPA, 4,4′-diisothiocyano-stilbene-2,2′-disulphonic acid (DIDS), 8-bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP), IAA, genistein, gentamicin, SIN-1, neomycin, nifedipine, ODQ, SNP (all from Sigma, St Louis, MO, USA), BAPTA-AM (from Dojindo, Osaka, Japan), cyclosporin A, forskolin, (±)-S-nitroso-N-acetylpenicillamine (SNAP; from Calbiochem, San Diego, CA, USA) and CGP-37157 (Tocris, Bristol, UK). CCCP, CGP-37157, CPA, forskolin, genistein, nifedipine and ODQ were dissolved in dimethyl sulphoxide (DMSO) to make stock solutions, and were added to Krebs solution to make the desired concentrations just prior to their use. Cyclosporin A was dissolved in ethanol. Other drugs tested were dissolved in distilled water. The final concentration of the solvent in Krebs solution did not exceed 1:1000. Addition of these chemicals to Krebs solution did not alter the pH of the solution.
Experimental data are expressed as the mean ± s.d. Statistical significance was tested using Student's t test, and probabilities of less than 5 % (P < 0.05) were considered significant.
Results
Effects of SNP on spontaneous electrical activities recorded from guinea pig antrum
In the presence of 1 μm nifedipine, three types of spontaneous electrical behaviour were recorded from the antral tissues of the guinea pig stomach; square-shaped potentials with slow (< 0.2 V s−1) rates of rise and subsequent plateau components with amplitudes of 20-30 mV (follower potentials), square-shaped potentials with fast (> 0.2 V s−1) rates of rise and subsequent plateau components with amplitudes of 40-50 mV (pacemaker potentials) and triangular potentials with slow (< 0.2 V s−1) rates of rise with amplitudes of 20-30 mV (slow waves). Comparison of the electrical properties of these potentials with those reported previously (Dickens et al. 1999, 2000; Hirst & Edwards, 2001) suggested that the slow waves, follower potentials and pacemaker potentials were recorded from circular muscle, longitudinal muscle and ICC-MY, respectively.
Experiments were carried out to test the effects of SNP on these electrical signals, all in the presence of 1 μm nifedipine. The effects were evaluated by measuring the peak amplitude, the frequency measured as a number of potentials within 1 min, and the duration of potentials measured at the half-amplitude of the peak (half-width). SNP (1-100 μm) increased the frequency and decreased the duration of pacemaker potentials, in a concentration-dependent manner (Fig. 1A-C; Table 1). These actions of SNP appeared with no alteration to the resting membrane potential and the amplitude or the maximum rate of rise (dV/dtmax) of pacemaker potentials (Fig. 1D, Table 1). SNP (100 μm) increased the frequency (control, 3.4 ± 0.7 min−1; in SNP, 6.6 ± 0.5 min−1; n = 7; P < 0.01; each n value represents a separate animal) and decreased the duration (the half-width in control, 8.4 ± 2.9 s; in SNP, 5.7 ± 1.8 s; n = 7; P < 0.05) of follower potentials, with no alteration to the amplitude (control, 30.9 ± 4.7 mV; in SNP, 30.2 ± 3.3 mV; n = 7; P > 0.05) and dV/dtmax (control, 0.13 ± 0.04 V s−1; in SNP, 0.15 ± 0.05 V s−1; n = 7; P > 0.05) (Fig. 2A). The resting membrane potential of follower-potential-generating cells was also not altered by SNP (control, −63.3 ± 3.0 mV; in SNP, −63.1 ± 2.2 mV; n = 7; P > 0.05). In the presence of 100 μm SNP, the frequency of slow waves was also increased (control, 3.5 ± 0.9 min−1; in SNP, 5.4 ± 0.7 min−1; n = 9; P < 0.01; Fig. 2B) and the amplitude of slow waves was decreased (control, 30.0 ± 5.0 mV; in SNP, 14.6 ± 5.8 mV; n = 9; P < 0.01). ODQ (10 μm), an inhibitor of guanylate cyclase, prevented the inhibitory actions of SNP on the amplitude of slow waves (control, 28.0 ± 4.5 mV; in ODQ, 28.5 ± 4.1 mV; n = 10; P > 0.05; in SNP with ODQ, 31.5 ± 3.6 mV; n = 10; P > 0.05), with no alteration to the resting membrane potential (control, −63.5 ± 2.8 mV; in ODQ, −63.4 ± 2.7 mV; n = 10; P > 0.05; in SNP with ODQ, −62.6 ± 2.8 mV; n = 10; P > 0.05) (Fig. 2C). However, ODQ did not alter the accelerating action of SNP on the frequency of slow waves (control, 3.2 ± 0.5 min−1; in ODQ, 3.2 ± 0.6 min−1; n = 10; P > 0.05; in SNP with ODQ, 5.7 ± 0.6 min−1; n = 10; P < 0.01). These results suggest that SNP inhibits the amplitude of slow waves in a cGMP-dependent manner, but increases the frequency of spontaneous activities through cGMP-independent mechanisms. The accelerating action of SNP on the frequency of pacemaker potentials was not altered in the presence of 1 μm atropine (n = 6) or 1 μm tetrodotoxin (n = 4, data not shown), suggesting that neuronal effects were not involved in these actions. The accelerating action of SNP on frequency continued for more than 20 min during application of SNP. The effects of SNP were reversible, and the activity reverted to the control condition by superfusion of tissues with SNP-free solution for 20-30 min (n = 5, data not shown).
Figure 1. Effects of sodium nitroprusside (SNP) on pacemaker potentials recorded from the guinea pig antrum.
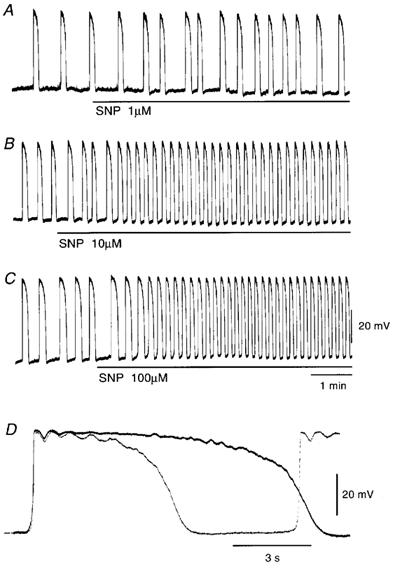
SNP (A, 1 μm; B, 10 μm; C, 100 μm) was applied as indicated by the bar under each record. D, high-speed trace of the pacemaker potential recorded in the absence (continuous line) and presence of 100 μm SNP (dotted line). The resting membrane potentials were: A, −63 mV; B, −62 mV; C and D, −64 mV. All responses were recorded from different tissues.
Table 1.
Effects of sodium nitroprusside (SNP) on the electrical properties of pacemaker potentials
| Membrane potential (mV) | Amplitude(primary, mV) | Amplitude(primary, mV) | Frequency(min−1) | Half-width(s) | dV/dtmax(V s−1) | n | |
|---|---|---|---|---|---|---|---|
| Control | −63.5 ± 3.8 | 47.0 ± 4.2 | 48.4 ± 4.2 | 3.1 ± 0.5 | 8.4 ± 1.7 | 0.66 ± 0.15 | 8 |
| SNP 1 μm | −63.5 ± 2.3 | 47.2 ± 4.6 | 48.6 ± 4.9 | 3.2 ± 0.5 | 8.4 ± 1.0 | 0.66 ± 0.16 | 8 |
| Control | −63.6 ± 3.0 | 45.7 ± 2.8 | 47.3 ± 3.4 | 3.2 ± 0.5 | 8.2 ± 1.2 | 0.54 ± 0.18 | 10 |
| SNP 10 μm | −63.7 ± 2.6 | 46.1 ± 3.0 | 47.6 ± 3.0 | 5.4 ± 0.8** | 5.7 ± 0.9** | 0.63 ± 0.11 | 10 |
| Control | −63.1 ± 2.3 | 49.5 ± 2.9 | 51.1 ± 3.6 | 3.2 ± 0.6 | 8.2 ± 1.4 | 0.54 ± 0.08 | 9 |
| SNP 100 μm | −62.7 ± 4.0 | 49.2 ± 4.1 | 50.4 ± 3.8 | 6.2 ± 0.8** | 5.6 ± 0.5** | 0.59 ± 0.10 | 9 |
Values are means ± s.d.n = number of tissues. *P < 0.05
P < 0.01.
Figure 2. Effects of SNP on follower potentials and slow waves recorded from guinea pig antrum.
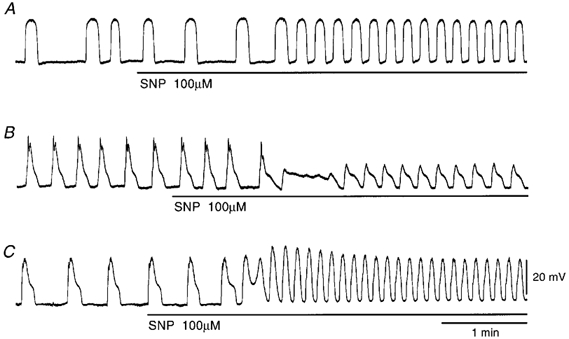
SNP (100 μm) was applied to follower potentials (A) and slow waves (B), as indicated by the bar under each record. C, SNP (100 μm) was applied to slow waves in the presence of 10 μm ODQ for 30 min. The resting membrane potentials were: A, −60 mV; B, −67 mV; C, −65 mV. All traces were recorded from different tissues.
Properties of SNP-induced pacemaker potentials
In the circular smooth muscles of the guinea pig stomach, forskolin, an activator of adenylate cyclase, inhibits spontaneous activity (Tsugeno et al. 1995). In the present study, forskolin (1 μm) inhibited the frequency and duration of pacemaker potentials with no alteration to the amplitude and dV/dtmax of the primary component (Table 2). In 14 cells examined, the membrane potentials were not significantly altered (Table 2). During the forskolin-induced inhibition of pacemaker potential generation, co-application of SNP (100 μm) again elicited the generation of pacemaker potentials (Fig. 3A and B). These actions of SNP on pacemaker potentials were not altered by 10 μm ODQ (n = 4, data not shown). A membrane-permeable analogue of cGMP, 8Br-cGMP (100 μm), exhibited no detectable effect on the generation of pacemaker potentials in the presence of 1 μm forskolin (n = 4, data not shown). The effects of two other types of NO donor, SNAP (100 μm) and SIN-1 (100 μm), were also tested, and no effect on the reappearance of pacemaker potentials during inhibition by 1 μm forskolin was observed (n = 8 for SNAP; n = 5 for SIN-1, data not shown). These results suggest that the excitatory effect of SNP upon forskolin-inhibited pacemaker potentials is independent of cGMP.
Table 2.
Effects of forskolin and 4,4′-diisothiocyano-stilbene-2,2′-disuphonic acid (DIDS) on the pacemaker potentials evoked by SNP
| Membrane potential (mV) | Amplitude (primary, mV) | Frequency (min−1) | Half-width (s) | dV/dtmax (V s−1) | n | |
|---|---|---|---|---|---|---|
| Control | −63.6 ± 3.3 | 45.8 ± 3.5 | 3.4 ± 0.7 | 7.9 ± 1.4 | 0.58 ± 0.07 | 14 |
| Forskolin 1 μm | −64.5 ± 3.7 | 45.8 ± 3.7 | 0.6 ± 0.8** | 0.6 ± 0.2** | 0.58 ± 0.08 | 14 |
| Forskolin 1 μm+ SNP 100 μm | −63.6 ± 3.2 | 45.8 ± 3.5 | 6.5 ± 1.9** | 0.6 ± 0.1** | 0.59 ± 0.08 | 14 |
| Control | −64.1 ± 4.2 | 51.3 ± 3.8 | 3.3 ± 0.7 | 7.8 ± 1.4 | 0.62 ± 0.10 | 12 |
| DIDS 300 μm | −65.5 ± 3.3 | 51.6 ± 3.6 | 1.9 ± 1.6** | 0.5 ± 0.3** | 0.44 ± 0.08 | 12 |
| DIDS 300 μm+ SNP 100 μm | −64.8 ± 3.9 | 51.1 ± 3.1 | 22.2 ± 2.2** | 0.4 ± 0.1** | 0.43 ± 0.06 | 12 |
Values are means ± s.d.n = number of tissues. *P < 0.05
P < 0.01.
Figure 3. Effects of SNP on pacemaker potentials in the presence of forskolin.
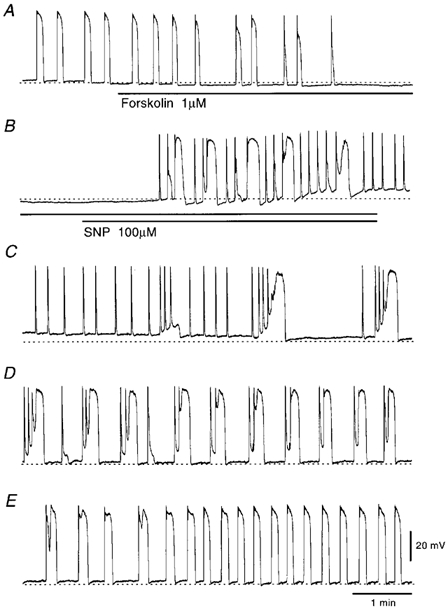
Pacemaker potentials recorded in response to 1 μm forskolin (A) and subsequent 100 μm SNP in addition to forskolin (B). C, D and E are responses of pacemaker potentials recorded during wash out of forskolin and SNP for 9 min, 24 min and 33 min, respectively. The resting membrane potential was −65 mV. All traces are continuous with some interruptions.
The effects of membrane depolarization on SNP-induced pacemaker potentials during forskolin inhibition were investigated. As shown in Fig. 4A, pacemaker potentials generated in the presence of forskolin and SNP were abolished during depolarization of the membrane (14.7 ± 0.9 mV, n = 5) with 15.3 mm [K+]o solution. In the presence of forskolin, the plateau component of pacemaker potentials was abolished and only the primary component remained (Fig. 4B and C). The SNP-elicited pacemaker potentials in the presence of forskolin appeared identical to those generated in the presence of forskolin alone (Fig. 4C and D). Comparison of pacemaker potentials generated in the presence of forskolin and forskolin with SNP showed no significant difference in amplitude, duration or dV/dtmax (Table 2). The SNP-induced pacemaker potentials were also abolished by application of low-Ca2+ solution ([Ca2+]o = 0.25 mm) in the presence of forskolin (n = 4, data not shown).
Figure 4. Effects of high [K+]o solution on SNP-evoked pacemaker potentials in the presence of forskolin.
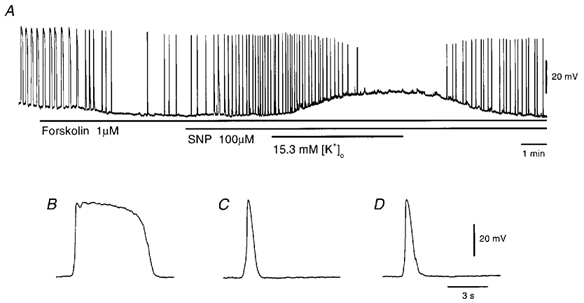
A, SNP-evoked pacemaker potentials were recorded during application of 15.3 mm [K+]o solution in the presence of 1 μm forskolin (applied as indicated by the bars under the record). High-speed traces of pacemaker potentials recorded before (B) and during application of 1 μm forskolin (C) and 1 μm forskolin with 100 μm SNP (D) are shown. All responses were recorded from the same cell with the resting membrane potential of −63 mV.
The possible involvement of Cl− channels on the SNP-generated pacemaker potentials was investigated by applying DIDS, an inhibitor of Ca2+-activated Cl− channels (Large et al. 1996), since this chemical inhibited the generation of spontaneous pacemaker potentials (Kito et al. 2002). In the presence of DIDS (300 μm), the frequency, duration and dV/dtmax of pacemaker potentials were decreased, with no significant alteration to their amplitude (Table 2). In 12 preparations tested, the mean value of the membrane potentials was not significantly different from that measured in the absence of DIDS (Table 2). In the presence of DIDS, SNP (100 μm) again elicited pacemaker-potential generation (Fig. 5A). The SNP-elicited pacemaker potentials were initially triangular forms (Fig. 5Bb), and with time they changed to a transient form with the primary component alone (Fig. 5Bc and Bd). The amplitude, duration and dV/dtmax of pacemaker potentials generated in the presence of DIDS were not further altered by addition of SNP (Table 2).
Figure 5. Effects of SNP on pacemaker potentials in the presence of 4,4′-diisothiocyano-stilbene-2,2′-disulphonic acid (DIDS).
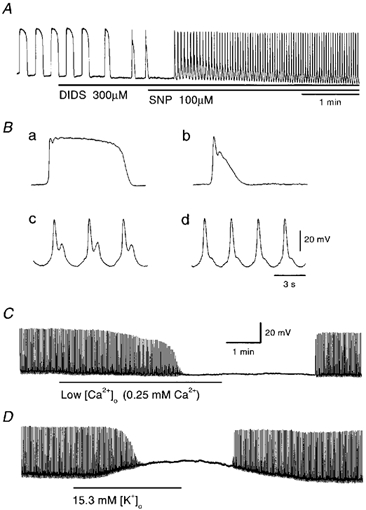
A, pacemaker potentials were recorded before and during application of DIDS (300 μm) followed by 100 μm SNP (applied as indicated by the bar under the record). High-speed traces of pacemaker potentials recorded before (Ba) and during application of 300 μm DIDS (Bb) and 300 μm DIDS with 100 μm SNP at 0.7 min (Bc) and 3 min (Bd) are shown. Low [Ca2+]o solution (C) and 15.3 mm [K+]o solution (D) were applied to SNP-induced pacemaker potentials in the presence of 300 μm DIDS (applied as indicated by the bar under the record). The resting membrane potentials were: A and B, −64 mV; C, −62 mV; D, −67 mV. All traces were recorded from different tissues.
In the presence of DIDS and SNP, the pacemaker potentials were abolished by low-Ca2+ solution ([Ca2+]o = 0.25 mm), with no significant alteration to the membrane potential (n = 6, Fig. 5C). The inhibition of pacemaker potentials by low-Ca2+ solution was reversible, with a quick recovery within 2-3 min. Depolarization of the membrane with 15.3 mm [K+]o solution abolished the pacemaker potential, with 16.5 ± 1.2 mV depolarization (n = 7, Fig. 5D). These results suggest that the primary component of pacemaker potentials is generated by the activation of voltage-dependent Ca2+-permeable channels.
Effects of Ni2+, Co2+ and aminoglycosides on pacemaker potentials
It has been demonstrated that both Ni2+ and Co2+ have potentiating actions on the slow waves recorded from guinea pig stomach (Tomita et al. 1998). In our preliminary experiments, it was found that neomycin increased the frequency of slow waves (Y. Kito, unpublished observation). Therefore, experiments were carried out to test the effects of Ni2+, Co2+ and aminoglycosides (neomycin and gentamicin) on pacemaker potentials. Ni2+ (10 μm) increased the frequency and reduced the duration of pacemaker potentials; similar actions were also elicited by Co2+ (10 μm; Fig. 6, Table 3). The reduction of the duration of pacemaker potential by Ni2+ or Co2+ was due mainly to inhibition of the plateau component (Fig. 6B and D). These effects of Ni2+ and Co2+ continued during the application (for more than 30 min). Recovery from the actions of Ni2+ and Co2+ was incomplete after 60 min of washout (n = 6).
Figure 6. Effects of Ni2+ and Co2+ on pacemaker potentials.
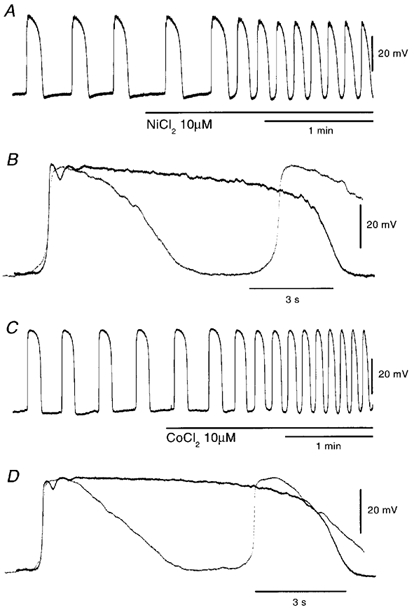
Ni2+ (10 μm) and Co2+ (10 μm) were applied to pacemaker potentials as indicated by the bar shown under records A and C, respectively. B and D are high-speed recordings of pacemaker potentials recorded in the absence (continuous lines) and presence of 10 μm Ni2+ or 10 μm Co2+ (dotted lines), respectively. The resting membrane potentials were: A and B, −62 mV; C and D, −62 mV. A and C were recorded from different tissues.
Table 3.
Effects of Ni2+, Co2+ and aminoglycosides on the properties of pacemaker potentials
| Membrane potential (mV) | Amplitude (primary, mV) | Amplitude (primary, mV) | Frequency (min−1) | Half-width (s) | dV/dtmax(V s−1) | n | |
|---|---|---|---|---|---|---|---|
| Control | −63.2 ± 4.1 | 47.7 ± 5.0 | 49.6 ± 4.1 | 3.2 ± 0.5 | 9.0 ± 0.7 | 0.58 ± 0.17 | 5 |
| Ni2+ 10 μm | −62.3 ± 3.6 | 47.1 ± 4.6 | — | 8.3 ± 0.7** | 3.5 ± 0.1** | 0.50 ± 0.13 | 5 |
| Control | −63.0 ± 3.1 | 47.4 ± 2.6 | 49.5 ± 2.2 | 3.3 ± 0.8 | 7.7 ± 2.2 | 0.63 ± 0.16 | 5 |
| Co2+ 10 μm | −61.7 ± 4.5 | 46.1 ± 4.2 | — | 8.4 ± 1.3** | 2.8 ± 0.4** | 0.56 ± 0.14 | 5 |
| Control | −64.1 ± 2.0 | 47.1 ± 4.5 | 48.6 ± 4.6 | 3.3 ± 0.7 | 8.4 ± 0.8 | 0.48 ± 0.02 | 5 |
| Neomycin 3 μm | −63.1 ± 2.4 | 46.6 ± 4.7 | 48.6 ± 5.2 | 5.8 ± 0.6** | 3.5 ± 1.0** | 0.50 ± 0.08 | 5 |
| Control | −64.3 ± 3.1 | 47.2 ± 3.6 | 48.8 ± 2.8 | 3.3 ± 0.5 | 8.2 ± 1.2 | 0.53 ± 0.08 | 4 |
| Gentamicin 3 μm | −64.4 ± 2.6 | 47.4 ± 3.9 | 48.6 ± 3.6 | 6.2 ± 0.7** | 3.9 ± 0.4* | 0.50 ± 0.09 | 4 |
Values are means ± s.d.n = number of tissues.
P < 0.05
P < 0.01.
In the guinea pig gastric antrum, both neomycin (3 mm) and gentamicin (3 mm) increased the frequency and decreased the duration of pacemaker potentials (Fig. 7A and B). These effects of aminoglycosides were not accompanied by changes in the resting potentials or dV/dtmax of the primary component of pacemaker potentials (Fig. 7C; Table 3), and were reversible within 30-40 min (n = 8).
Figure 7. Effects of aminoglycosides on pacemaker potentials.
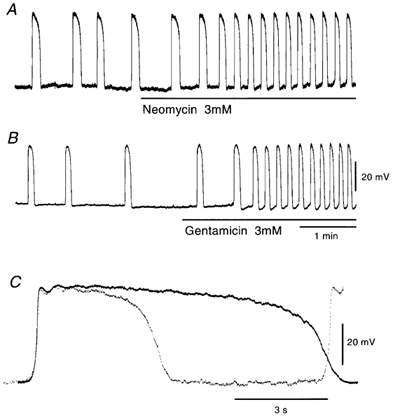
Pacemaker potentials were recorded during application of 3 mm neomycin (A) or 3 mm gentamicin (B). C, high-speed recordings of pacemaker potentials, in the absence (continuous lines) and presence of 3 mm neomycin (dotted lines). The resting membrane potentials were: A and C, −64 mV; B, −61 mV. A and B were recorded from different tissues
The effects of Ni2+, Co2+ and aminoglycosides were observed in tissues whose activities had been inhibited with forskolin. As shown in Fig. 8A, Ni2+ (10 μm) elicited generation of pacemaker potentials in the presence of 1 μm forskolin, with no alteration to the resting potential (control, −61.7 ± 2.0 mV; in forskolin, −62.2 ± 2.0 mV; n = 4; P > 0.05; in Ni2+ with forskolin, −61.2 ± 2.2 mV; n = 4; P > 0.05). The pacemaker potentials elicited by Ni2+ were similar in amplitude (control, 42.3 ± 3.2 mV; in forskolin, 41.2 ± 1.2 mV; n = 4; P > 0.05; in Ni2+ with forskolin, 42.9 ± 5.2 mV; n = 4; P > 0.05) and dV/dtmax (control, 0.48 ± 0.15 V s−1; in forskolin, 0.54 ± 0.17 V s−1; n = 4; P > 0.05; in Ni2+ with forskolin, 0.46 ± 0.14 V s−1; n = 4; P > 0.05) with those generated in the absence of forskolin or in the presence of forskolin without Ni2+. However, their duration was much longer than that in the absence of forskolin (half-width in control, 7.8 ± 1.4 s; in forskolin with Ni2+, 15.9 ± 4.1 s; n = 4; P < 0.01). Co2+ (10 μm) applied in the presence of forskolin elicited the initial component of a pacemaker potential and subsequent depolarization, which recovered slowly to the resting level within 2-3 min, with sustained generation of pacemaker potentials displaying long duration (Fig. 8B). In five tissues examined, Co2+ did not alter the membrane potential (control, −62.2 ± 3.1 mV; in forskolin, −62.6 ± 2.2 mV; P > 0.05; in Co2+ with forskolin, −62.0 ± 2.4 mV; P > 0.05), amplitude (control, 47.6 ± 4.8 mV; in forskolin, 46.2 ± 5.4 mV; P > 0.05; in Co2+ with forskolin, 47.0 ± 4.6 mV; P > 0.05) nor dV/dtmax (control, 0.57 ± 0.24 V s−1; in forskolin, 0.56 ± 0.22 V s−1; P > 0.05; in Co2+ with forskolin, 0.49 ± 0.17 V s−1; P > 0.05) of pacemaker potentials.
Figure 8. Effects of Ni2+, Co2+ and neomycin on pacemaker potentials in the presence of forskolin.
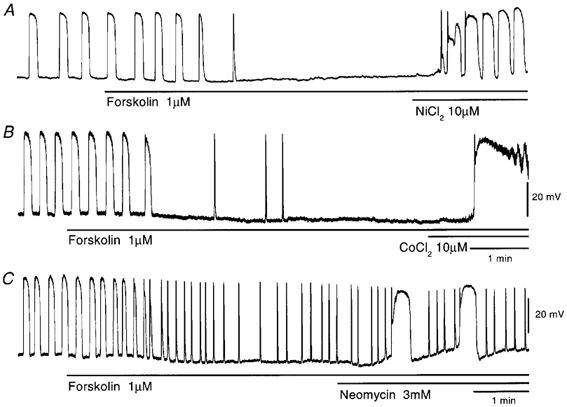
Pacemaker potentials in response to 10 μm Ni2+ (A), 10 μm Co2+ (B) or 3 mm neomycin (C) (applied at the bar under each record) were recorded in the presence of 1 μm forskolin. The resting membrane potentials were: A, −63 mV; B, −66 mV; C, −64 mV. All traces were recorded from different tissues.
Neomycin (3 mm) applied in the presence of forskolin produced pacemaker potentials with plateau components (Fig. 8C). The membrane potential (control, −62.7 ± 2.7 mV; in forskolin, −63.6 ± 2.0 mV; n = 6; P > 0.05; in neomycin with forskolin, −62.4 ± 1.6 mV; n = 6; P > 0.05), amplitude of the primary component (control, 45.3 ± 5.7 mV; in forskolin, 44.7 ± 5.7 mV; n = 6; P > 0.05; in neomycin with forskolin, 45.1 ± 5.7 mV; n = 6; P > 0.05) and dV/dtmax of pacemaker potentials (control, 0.50 ± 0.08 V s−1; in forskolin, 0.49 ± 0.08 V s−1; n = 6; P > 0.05; in neomycin with forskolin, 0.45 ± 0.10 V s−1; n = 6; P > 0.05) were not altered by neomycin. Thus, Ni2+, Co2+ and aminoglycosides have similar effects on the pacemaker potentials generated in the presence of forskolin, and these actions were also similar to those of SNP (Fig. 3).
Effects of genistein on SNP-induced generation of pacemaker potentials
The effects of genistein, a tyrosine kinase inhibitor, on the SNP-induced generation of pacemaker potentials were investigated, since the SNP-induced contractions are inhibited by this tyrosine kinase inhibitor in opossum oesophageal longitudinal smooth muscle (Hirano et al. 1997) and guinea pig gall bladder smooth muscle (Alcon et al. 2001). In the presence of both 1 μm forskolin and 30 μm genistein for 15-20 min, SNP (100 μm) generated pacemaker potentials as in the case with the absence of genistein (n = 4, data not shown). These results suggest that tyrosine kinase activity is unlikely to be required for the SNP-induced generation of pacemaker potentials.
Role of intracellular Ca2+ in the SNP-induced generation of pacemaker potentials
The above observations indicated that SNP increased the amplitude of slow waves in the presence of ODQ (Fig. 2C). The second component of slow waves is generated by the opening of Ca2+-activated Cl− channels (Hirst et al. 2002). Furthermore, the SNP-induced primary component in the presence of forskolin or DIDS was abolished by application of low-Ca2+ solution (Fig. 5C). These results raise the possibility that SNP exerts its excitatory effects on pacemaker potentials by increasing intracellular Ca2+ concentrations. The possible contribution of internal Ca2+ stores on the SNP-induced generation of pacemaker potentials was studied in the presence of CPA, an inhibitor of Ca2+-ATPase at the internal membrane (Uyama et al. 1992). CPA (10 μm) depolarized the membrane (6.6 ± 3.1 mV, n = 5) and increased the frequency (control, 3.5 ± 0.9 min−1; in CPA, 4.8 ± 0.6 min−1, n = 5; P < 0.05) and decreased the duration of pacemaker potentials (half-width in control, 8.9 ± 1.3 s; in CPA, 3.8 ± 0.6 s; n = 5; P < 0.01), with no alteration to dV/dtmax (control, 0.57 ± 0.17 V s−1; in CPA, 0.55 ± 0.16 V s−1; n = 5; P > 0.05), as had been reported previously (Kito et al. 2002). In the presence of 10 μm CPA for over 20 min, SNP (100 μm) increased the frequency of pacemaker potentials (in CPA, 4.8 ± 0.6 min−1; in SNP with CPA, 6.6 ± 1.7 min−1, n = 5; P < 0.05), with no alteration to the membrane potential (in CPA, −56.5 ± 3.2 mV, in SNP with CPA, −56.9 ± 2.7 mV; n = 5; P > 0.05) or dV/dtmax (in CPA, 0.55 ± 0.16 V s−1; in SNP with CPA, 0.56 ± 0.18 V s−1; n = 5; P > 0.05) of pacemaker potentials (Fig. 9A). These results suggest that the SNP-induced generation of pacemaker potentials is not related to Ca2+ released from the internal stores.
Figure 9. Effects of SNP on pacemaker potentials in the presence of cyclopiazonic acid (CPA) or 1,2-bis(2-aminophenoxy)ethane-N,N,N‘,N‘-tetraacetic acid acetoxymethyl ester (BAPTA-AM).
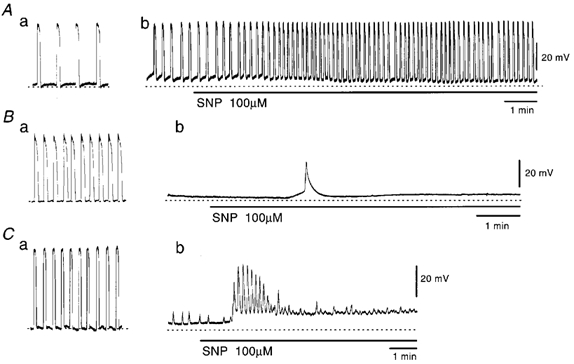
Pacemaker potentials were recorded before (a) and during (b) application of 10 μm CPA for 30 min (A), 50 μm BAPTA-AM for 30 min (B) and 50 μm BAPTA-AM at 60 min (C). In the presence of these chemicals for 30 min (A and B) or 60 min (C), 100 μm SNP was applied at the bar under each record. The resting membrane potentials shown by dotted lines are: A, −63 mV; B, −60 mV; C, −66 mV. All traces were recorded from different tissues.
The effects of BAPTA-AM, an intracellular Ca2+ chelator, on the SNP-induced generation of pacemaker potentials were also studied. BAPTA-AM (50 μm) abolished pacemaker potentials, with depolarization of the membrane by 4.2 ± 2.0 mV (n = 18). In the presence of BAPTA-AM, SNP (100 μm) elicited a transient burst of pacemaker potentials. The evoked pacemaker potentials were solitary (Fig. 9B) or repetitive (Fig. 9C), occurring between 1 and 56 times (mean, 16.3 ± 14.0 times; n = 18). The pacemaker potentials elicited by SNP in the presence of BAPTA-AM were smaller in amplitude (control, 49.7 ± 4.2 mV, in SNP with BAPTA-AM, 31.6 ± 4.2 mV; n = 18; P < 0.01) and dV/dtmax (control, 0.57 ± 0.12 V s−1; in SNP with BAPTA-AM, 0.12 ± 0.08 V s−1; n = 18; P < 0.01). These results suggest that the SNP-induced increase in the generation of pacemaker potentials is related to the elevation of intracellular Ca2+ concentration, and independent of the functions of internal Ca2+ stores.
Role of mitochondrial functions on the SNP-induced generation of pacemaker potentials
The effects of impairment of mitochondrial functions on the SNP-induced generation of pacemaker potentials were examined. CCCP (3 μm), a mitochondrial protonophore (Gunter & Pfeiffer, 1990), depolarized the membrane by 12.5 ± 3.1 mV and abolished pacemaker potentials (n = 11). After pacemaker potentials had been abolished with CCCP, SNP (100 μm) again generated pacemaker potentials, but only transiently (Fig. 10A). The number of evoked pacemaker potentials ranged between 1 and 24 (mean, 11.5 ± 7.6 times; n = 11). The amplitude (control, 48.8 ± 5.7 mV, in SNP with CCCP, 23.1 ± 8.1 mV; n = 11; P < 0.01) and dV/dtma (control, 0.59 ± 0.11 V s−1; in SNP with CCCP, 0.18 ± 0.18 V s−1; n = 11; P < 0.01) of the evoked pacemaker potentials were significantly smaller than in the absence of CCCP. In five out of 11 preparations, SNP (100 μm) transiently repolarized the membrane by 3.5 ± 1.0 mV.
Figure 10. Effects of SNP on pacemaker potentials recorded in the presence of carbonyl cyanide m-chlorophenyl-hydrazone (CCCP) and iodoacetic acid (IAA).
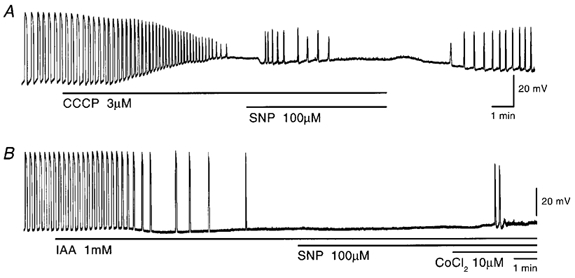
Pacemaker potentials were recorded before and during application of 3 μm CCCP (A) or 1 mm IAA (B), and 100 μm SNP was added as indicated by the bar under the record. In B, 10 μm Co2+ was also added together with IAA and SNP. The resting membrane potentials were: A, −62 mV; B, −65 mV. A and B were recorded from different tissues.
IAA (1 mm), an inhibitor of glycolysis (Ogino et al. 1999), transiently hyperpolarized the membrane by 2.9 ± 1.1 mV (n = 9) and abolished pacemaker potentials (Fig. 10B). In the presence of IAA, SNP (100 μm) elicited a transient burst of pacemaker potentials, the number ranging between 0 and 4 (mean, 0.8 ± 1.3 times; n = 9). The evoked pacemaker potentials were identical in amplitude (control, 50.7 ± 6.4 mV, in SNP with IAA, 41.2 ± 6.5 mV; n = 9; P > 0.05) and dV/dtma (control, 0.56 ± 0.16 V s−1; in SNP with IAA 0.48 ± 0.08 V s−1; n = 9; P > 0.05). In the presence of IAA and SNP, additional co-application of Co2+ (10 μm) transiently generated pacemaker potentials, the number ranging between 2 and 33 (mean, 12.8 ± 9.6 times; n = 8; Fig. 10B). Taken together, these results suggest that mitochondrial functions are important for the SNP-induced generation of pacemaker potentials.
Effects of inhibition of the extrusion of Ca2+ from mitochondria on the SNP-induced generation of slow waves
The extrusion of Ca2+ from mitochondria is mediated through the Na+/Ca2+ exchanger or mitochondrial permeability transition pore (MTP; Bernardi, 1999). Experiments were carried out to investigate the effects of CGP-37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger (Kaftan et al. 2000; Arnaudeau et al. 2001), and cyclosporin A, a blocker of MTP (Bernardi, 1999), on the SNP-induced generation of slow waves. Application of CGP-37157 (10 μm) for 30-40 min did not prevent the SNP-induced generation of slow waves. The membrane potential (control, −65.0 ± 4.0 mV; in CGP-37157, −64.9 ± 3.2 mV; n = 8; P > 0.05; in SNP with CGP-37157, −64.3 ± 3.5 mV; n = 8; P > 0.05), and amplitude (control, 27.3 ± 1.3 mV; in CGP-37157, 27.7 ± 1.6 mV; n = 8; P > 0.05; in SNP with CGP-37157, 16.0 ± 2.5 mV; n = 8; P < 0.05) and frequency (control, 3.2 ± 0.4 min−1; in CGP-37157, 3.1 ± 0.8 min−1; n = 8; P > 0.05; in SNP with CGP-37157, 6.3 ± 1.3 min−1; n = 8; P < 0.05) of slow waves were also not altered by CGP-37157. The actions of SNP on the generation of slow waves were again not altered in the presence of cyclosporin A (5 μm) for 30-40 min. The membrane potential (control, −65.6 ± 3.9 mV; in cyclosporin A, −65.5 ± 4.6 mV; n = 8; P > 0.05; in SNP with cyclosporin A, −66.0 ± 4.6 mV; n = 8; P > 0.05), and amplitude (control, 28.3 ± 4.9 mV; in cyclosporin A, 27.5 ± 4.9 mV; n = 8; P > 0.05; in SNP with cyclosporin A, 13.9 ± 3.5 mV; n = 8; P < 0.05) and frequency (control, 3.2 ± 0.7 min−1; in cyclosporin A, 4.0 ± 0.8 min−1; n = 8; P > 0.05; in SNP with cyclosporin A, 6.4 ± 1.3 min−1; n = 8; P < 0.05) of slow waves were not altered by cyclosporin A. These results suggest that neither the mitochondrial Na+/Ca2+ exchanger nor MTP is involved in the SNP-induced generation of slow waves.
Discussion
The experiments presented here demonstrated that in the gastric antrum, SNP increases the frequency of pacemaker potentials, follower potentials and slow waves, with no alteration to the amplitude of pacemaker and follower potentials, but with reduction in the amplitude of slow waves. The SNP-induced inhibition of the amplitude of slow waves was prevented by ODQ, an inhibitor of guanylate cyclase, suggesting that the inhibition is related to the elevated production of cGMP through activation of guanylate cyclase. In the isolated circular smooth muscles of the guinea pig antrum with no attached longitudinal muscle layer or ICC-MY, slow potentials are generated periodically and these potentials are thought to be the second component of slow waves (Tomita, 1981), since they are inhibited by low concentrations of caffeine (Suzuki & Hirst, 1999; Edwards et al. 1999; Fukuta et al. 2002). SNP (1 μm) is also a potent inhibitor of slow potentials (Y. Kito, unpublished observations). Thus, the reduction in amplitude of slow waves by SNP may be due mainly to the inhibition of the second component of slow waves. The present experiments indicate further that this SNP-induced inhibition is produced in a cGMP-dependent manner. The excitatory actions of SNP on pacemaker and follower potentials were, on the other hand, not altered by ODQ, suggesting that the actions do not involve cGMP production. These results suggest an explanation for recent findings that in the guinea pig stomach muscles, vagal stimulation evokes NO-mediated inhibitory junction potentials that might interrupt the regenerative component of slow waves but not the driving and follower potentials (Dickens et al. 2000). ICC-MY may be electrically coupled to each other through gap junctions, and similar pathways are also considered for the transmission of signals from ICC-MY to smooth muscles (Komuro et al. 1996; Huizinga et al. 1997; Sanders et al. 1999). Therefore, it seems likely that the cGMP-independent actions of SNP are initiated in ICC-MY and are conducted to the circular and longitudinal smooth muscles through gap junctions.
Forskolin inhibits the generation of spontaneous activity in gastric muscles, possibly due to elevated production of cAMP (Tsugeno et al. 1995; Kito et al. 2002). The present experiments indicate further that the inhibition affects mainly the frequency and plateau component of pacemaker potentials. In the presence of forskolin, SNP again elicited the generation of pacemaker potentials. This excitatory action of SNP was not altered by ODQ and was not mimicked by 8Br-cGMP or other types of NO donor (SIN-1, SNAP). These results again suggest that the excitatory action on pacemaker potentials by SNP is not due to the increase in production of cGMP, rather it may be related to its chemical nature other than as an NO donor. In the presence of forskolin, the pacemaker potentials generated by SNP consisted mainly of the primary component alone, since the plateau component was inhibited by forskolin. However, pacemaker potentials with long plateau components were also often generated in the presence of forskolin and SNP (Fig. 3). The mechanisms underlying the generation of the plateau component in the presence of both forskolin and SNP remain unclear.
The pacemaker potentials evoked by SNP were abolished either in low-Ca2+ solution or by depolarization with high-K+ solution. These results suggest that voltage-dependent Ca2+-permeable channels are involved in generating the primary component of pacemaker potentials. This suggestion is in good agreement with our recent findings that depolarization of the membrane with high [K+]o solutions reduced the dV/dtmax of pacemaker potentials (Kito et al. 2002). Forskolin does not alter the amplitude or dV/dtmax of pacemaker potentials, and these parameters are also unaltered for the pacemaker potentials evoked by SNP. Thus, SNP promotes the generation of the primary component of pacemaker potentials. Interestingly, in the process of recovery from the actions of forskolin and SNP, the plateau component developed successively with repetitive firing of primary components (Fig. 3). It may be that partial refilling of internal Ca2+ stores during the recovery process from forskolin-induced inhibition allows the generation of plateau components due to the repetitive release of Ca2+ from internal stores. The duration of the primary component is about 1 s, a time comparable to the delay required for the production of inositol 1,4,5-trisphosphate (IP3) in response to stimulation with agonists (Somlyo & Somlyo, 1994). These results support the idea that the depolarization induced by the primary component increases IP3 production and stimulates the release of Ca2+ from internal stores to generate the plateau component (Hirst & Edwards, 2001).
The plateau component of pacemaker potentials is absent in the presence of DIDS, suggesting that this component is formed by the opening of Ca2+-activated Cl− channels. In the presence of DIDS, SNP again evoked pacemaker potentials with frequencies higher than in the absence of DIDS. These results suggest a causal relationship between the duration of pacemaker potentials and their frequency. Although the amplitude, duration and dV/dtmax of the SNP-evoked pacemaker potentials remained unaltered, they were abolished in low-Ca2+ solution or during depolarization with high-K+ solution. Thus, the pacemaker potentials evoked by SNP in the presence of DIDS are similar to those evoked in the presence of forskolin. However, the pacemaker potentials evoked by SNP in the presence of DIDS are accompanied by slow diastolic phases, as in the case of cardiac pacemaker potentials recorded from sinoatrial node (Mitsuiye et al. 2000). The mechanisms underlying the generation of pacemaker potentials with diastolic phases by SNP in the presence of DIDS remain unclear.
SNP continued to evoke pacemaker potentials after the internal Ca2+ pump had been inhibited with CPA, suggesting that the release of Ca2+ from the internal stores is not the essential factor involved. As SNP increased the amplitude of second component of slow waves in the presence of ODQ and pacemaker potentials were inhibited in low-Ca2+ solutions, the actions of SNP may be related to an elevation of intracellular Ca2+ concentration. SNP elicits only a transient burst of pacemaker potentials in the presence of BAPTA-AM. The SNP-induced generation of pacemaker potentials was limited to a transient burst after mitochondrial Ca2+ handling had been disordered with CCCP, a mitochondrial protonophore, or with IAA, a glycolysis inhibitor. These results suggest that the mitochondrial Ca2+ functions are involved in the SNP-induced generation of pacemaker potentials. In many types of cell, CCCP diminishes mitochondrial Ca2+ uptake by reducing the potential difference across the inner membrane, thus elevating the outflow of mitochondrial Ca2+ (Gunter & Pfeiffer, 1990; White & Reynolds, 1997; Sorimachi et al. 1999; Murchison & Griffith, 2000; Montero et al. 2001). As a consequence, CCCP would reduce the amount of mitochondrial Ca2+ available due to the inhibition of uptake and facilitation of the release of Ca2+. Chelating intracellular Ca2+ with BAPTA-AM also reduces the available Ca2+ in mitochondria by decreasing uptake. In some preparations, pacemaker potentials were not abolished by application of 50 μm BAPTA-AM for about 1 h (Fig. 9Cb; Kito et al. 2002). These results suggest that there are some BAPTA-AM-resistant local spaces between mitochondrial membrane and the plasma membrane. It may be that even in the presence of BAPTA-AM, intracellular Ca2+ handling mechanisms still work and SNP activates these mechanisms by releasing Ca2+ from mitochondria to these discrete subcellular domains. Taken together, SNP may cause the release of Ca2+ from mitochondria in pacemaker cells and consequently accelerate the frequency of pacemaker potentials.
The results of the present study also show that Ni2+, Co2+ and aminoglycosides can mimic the actions of SNP on pacemaker potentials (i.e. these chemicals evoked or increased the frequency of pacemaker potentials after their generation had been abolished by forskolin). However, the difference between SNP and these agents appeared in the duration of the evoked pacemaker potentials, and the plateau component was restored by the latter in the presence of forskolin. It remains unclear what mechanisms are involved in the generation of the plateau component. In several types of cells, Ni2+, Co2+ and aminoglycosides are reported to modulate mitochondrial functions (Rubanyi & Balogh, 1982; Bragadin & Viola, 1997; Rustenbeck et al. 1998; Kakinuma et al. 2000; Mather & Rottenberg, 2001), and it is suggested that this is also the case for gastric pacemaker cells.
Intracellular Ca2+ homeostasis is maintained by mitochondrial Ca2+ handling, together with internal Ca2+ stores, and two common pathways have been identified for the efflux of Ca2+ from mitochondria: a Na+/Ca2+ exchanger and mitochondrial permeability transition pores (MPTs; Bernardi, 1999). CGP-37157, a selective inhibitor of the mitochondrial Na+/Ca2+ exchanger, blocks the efflux of Ca2+ from mitochondria in HeLa cells or rat gonadotropes (Kaftan et al. 2000; Arnaudeau et al. 2001). Cyclosporin A, an inhibitor of MPT, also inhibits the release of Ca2+ from mitochondria in rat forebrain neurons (Murchison & Griffith, 2000). The present experiments indicate that neither CGP-37157 nor cyclosporin A can prevent the SNP-induced generation of pacemaker activities, suggesting that neither the Na+/Ca2+ exchanger nor the MPT are involved in the actions of SNP. Involvement of other extrusion mechanisms (e.g. the H+/Ca2+ exchanger; Bernardi, 1999) in the actions of SNP on pacemaker potentials cannot be ruled out, and further experiments are required.
Mitochondria are in close contact with Ca2+-release sites in the endoplasmic reticulum (Rizzuto et al. 1998; Hajnoczky et al. 2000), and during Ca2+ oscillation, Ca2+ released from the endoplasmic reticulum is taken up into mitochondria and subsequently released to the cytosol (Kaftan et al. 2000; Arnaudeau et al. 2001). It is thought that residual Ca2+ not involved in mitochondrial Ca2+ handling activates a set of Ca2+-dependent ion channels at the plasma membrane to form unitary potentials. Possible involvement of Ca2+-activated Cl− channels in the generation of slow potentials (Hirst et al. 2002) or plateau component of pacemaker potentials (Kito et al. 2002) has been reported. Taken together, the following processes are considered to contribute to the generation of pacemaker potentials in ICC-MY: (1) Ca2+ released from mitochondria activates Ca2+-activated Cl− channels to generate unitary potentials; (2) when the potentials exceed the threshold level for excitation, voltage-gated Ca2+-permeable channels are opened to form the primary component of pacemaker potentials; (3) this depolarization elicits an influx of Ca2+ and elevates the production of IP3 through the activation of a Ca2+-sensitive enzyme such as phospholipase Cδ (Rebecchi & Pentyala, 2000); (4) elevated IP3 causes the release of Ca2+ from internal stores and generates the plateau component by opening Ca2+-activated Cl− channels. In these processes, SNP would increase the generation of unitary potentials by elevating the release of Ca2+ from mitochondria and thus facilitate the generation of the primary component of pacemaker potentials.
It is concluded that in the guinea pig gastric antrum, SNP increases the frequency of spontaneous activity by releasing Ca2+ from mitochondria in pacemaker cells. These actions of SNP are elicited independent of the production of cGMP. SNP also works as an NO donor, and the released NO inhibits generation of the second component of slow waves, inhibiting gastric contractions. Since SNP plays an integral role in the orientation or activation of the pacemaker activity, and since SNP-induced regenerative effects appear rapidly and reversibly, SNP seems to be a useful tool with which to elucidate the mechanisms underlying the pacemaker activity of ICC-MY in guinea pig antrum.
Acknowledgments
The authors are grateful to Dr Frank R. Edwards for critical reading of the manuscript. The present experiments were supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan to H.S. (No. 14570044).
References
- Alcon S, Morales S, Camello PJ, Hemming JM, Jennings L, Mawe GM, Pozo M. A redox-based mechanism for the contractile and relaxing effects of NO in the guinea-pig gall bladder. J Physiol. 2001;532:793–810. doi: 10.1111/j.1469-7793.2001.0793e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudeau S, Kelley WL, Walsh JV, Jr, Demaurex N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bragadin M, Viola ER. Ni2+ as a competitive inhibitor of calcium transport in mitochondria. J Inorg Biochem. 1997;66:227–229. doi: 10.1016/s0162-0134(96)00207-3. [DOI] [PubMed] [Google Scholar]
- Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol. 2000;184:409–420. doi: 10.1002/1097-4652(200009)184:3<409::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Dickens EJ, Edwards FR, Hirst GDS. Vagal inhibition in the antral region of guinea pig stomach. Am J Physiol. 2000;279:G388–399. doi: 10.1152/ajpgi.2000.279.2.G388. [DOI] [PubMed] [Google Scholar]
- Dickens EJ, Hirst GDS, Tomita T. Identification of rhythmically active cells in guinea-pig stomach. J Physiol. 1999;514:515–531. doi: 10.1111/j.1469-7793.1999.515ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FR, Hirst GDS, Suzuki H. Unitary nature of regenerative potentials recorded from circular smooth muscle of guinea-pig antrum. J Physiol. 1999;519:235–250. doi: 10.1111/j.1469-7793.1999.0235o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuta H, Kito Y, Suzuki H. Spontaneous electrical activity and associated changes in calcium concentration in guinea-pig gastric smooth muscle. J Physiol. 2002;540:249–260. doi: 10.1113/jphysiol.2001.013306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P. The machinery of local Ca2+ signaling between sarco-endoplasmic reticulum and mitochondria. J Physiol. 2000;529:69–81. doi: 10.1111/j.1469-7793.2000.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano I, Kakkar R, Saha JK, Szymanski PT, Goyal RK. Tyrosine phosphorylation in contraction of opossum esophageal longitudinal muscle in response to SNP. Am J Physiol. 1997;273:G247–252. doi: 10.1152/ajpgi.1997.273.1.G247. [DOI] [PubMed] [Google Scholar]
- Hirst GDS, Bramich NJ, Teramoto N, Suzuki H, Edwards FR. Regenerative component of slow waves in the guinea pig gastric antrum involves a delayed increase in [Ca2+]i and Cl− channels. J Physiol. 2002;540:907–919. doi: 10.1113/jphysiol.2001.014803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst GDS, Edwards FR. Generation of slow waves in the antral region of guinea-pig stomach - a stochastic process. J Physiol. 2001;535:165–180. doi: 10.1111/j.1469-7793.2001.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, Lippe IT, Tabrizi AL, Lenard L, Jr, Bartho L. Dual excitatory and inhibitory effect of nitric oxide on peristalsis in the guinea pig intestine. J Pharmacol Exp Ther. 1997;280:154–161. [PubMed] [Google Scholar]
- Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/Kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–349. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- Huizinga JD, Thuneberg L, Vanderwinden J-M, Rumessen J. Interstitial cells of Cajal as targets for pharmacological intervention in gastrointestinal motor disorders. Trends Pharm Sci. 1997;18:393–403. doi: 10.1016/s0165-6147(97)01108-5. [DOI] [PubMed] [Google Scholar]
- Kaftan EJ, Xu T, Abercrombie RF, Hille B. Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J Biol Chem. 2000;275:25465–25470. doi: 10.1074/jbc.M000903200. [DOI] [PubMed] [Google Scholar]
- Kakinuma Y, Miyauchi T, Yuki K, Murakoshi N, Goto K, Yamaguchi I. Mitochondrial dysfunction of cardiomyocytes causing impairment of cellular energy metabolism induces apoptosis, and concomitant increase in cardiac endothelin-1 expression. J Cardiovasc Pharm. 2000;36:S201–204. doi: 10.1097/00005344-200036051-00061. [DOI] [PubMed] [Google Scholar]
- Kito Y, Fukuta H, Suzuki H. Components of pacemaker potentials recorded from the guinea-pig stomach antrum. Pflugers Arch. 2002 doi: 10.1007/s00424-002-0884-z. DOI 10.1007/s00424-002-0884z. [DOI] [PubMed] [Google Scholar]
- Komuro T, Tokui K, Zhou DS. Identification of the interstitial cells of Cajal. Histol Histopathol. 1996;11:769–786. [PubMed] [Google Scholar]
- Kuriyama H, Kitamura K, Itoh T, Inoue R. Physiological features of visceral smooth muscle cells, with special reference to receptors and ion channels. Physiol Rev. 1998;78:811–920. doi: 10.1152/physrev.1998.78.3.811. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am J Physiol. 1996;271:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Lefebvre RA, Bartho L. Mechanism of nitric oxide-induced contraction in the rat isolated small intestine. Br J Pharm. 1997;120:975–981. doi: 10.1038/sj.bjp.0700996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J. 1993;7:328–338. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K, Nishikawa S. Requirement of c-kit for development of intestinal pacemaker system. Development. 1992;116:369–375. doi: 10.1242/dev.116.2.369. [DOI] [PubMed] [Google Scholar]
- Mather M, Rottenberg H. Polycations induce the release of soluble intermembrane mitochondrial proteins. Biochem Biophys Acta. 2001;1503:357–368. doi: 10.1016/s0005-2728(00)00231-0. [DOI] [PubMed] [Google Scholar]
- Mitsuiye T, Shinagawa Y, Noma A. Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Circ Res. 2000;87:88–91. doi: 10.1161/01.res.87.2.88. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, Garcia-Sancho J, Alvarez J. Mitochondrial Ca2+-induced Ca2+ release mediated by the Ca2+ uniporter. Mol Biol Cell. 2001;12:63–71. doi: 10.1091/mbc.12.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison D, Griffith WH. Mitochondria buffer non-toxic calcium loads and release calcium through the mitochondrial permeability transition pore and sodium/calcium exchanger in rat basal forebrain neurons. Brain Res. 2000;854:139–151. doi: 10.1016/s0006-8993(99)02297-0. [DOI] [PubMed] [Google Scholar]
- Ogino K, Takai A, Ishida Y, Tomita T. Effects of iodoacetate on spontaneous electrical activity, slow wave, in the circular muscle of the guinea-pig gastric antrum. Jpn J Physiol. 1999;49:521–526. doi: 10.2170/jjphysiol.49.521. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rubanyi G, Balogh I. Effect of nickel on uterine contraction and ultrastructure in the rat. Am J Obstet Gynecol. 1982;142:1016–1020. doi: 10.1016/0002-9378(82)90785-2. [DOI] [PubMed] [Google Scholar]
- Rustenbeck I, Eggers G, Reiter H, Munster W, Lenzen S. Polyamine modulation of mitochondrial calcium transport. I. Stimulatory and inhibitory effects of aliphatic polyamines, aminoglycosides and other polyamine analogues on mitochondrial calcium uptake. Biochem Pharm. 1998;56:977–985. doi: 10.1016/s0006-2952(98)00232-9. [DOI] [PubMed] [Google Scholar]
- Sanders KM, Ördög T, Koh SD, Torihashi S, Ward SM. Development and plasticity of interstitial cells of Cajal. Neurogastroenterol Motil. 1999;11:311–338. doi: 10.1046/j.1365-2982.1999.00164.x. [DOI] [PubMed] [Google Scholar]
- Sanders KM, Ward SM. Nitric oxide as a mediator of nonadrenergic noncholinergic neurotransmission. Am J Physiol. 1992;262:G379–392. doi: 10.1152/ajpgi.1992.262.3.G379. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Sorimachi M, Nishimura S, Yamagami K. Sequestration of depolarization-induced Ca2+ loads by mitochondria and Ca2+ efflux via mitochondrial Na+/Ca2+ exchanger in bovine adrenal chromaffin cells. Jpn J Physiol. 1999;49:35–46. doi: 10.2170/jjphysiol.49.35. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Hirst GDS. Regenerative potentials evoked in circular smooth muscle of the antral region of guinea-pig stomach. J Physiol. 1999;517:563–573. doi: 10.1111/j.1469-7793.1999.0563t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T. Electrical activity (spikes and slow wave) in gastrointestinal smooth muscles. In: B¨lbring E, Brading AF, Jones AW, Tomita T, editors. Smooth Muscle. London: Edward Arnold; 1981. pp. 127–156. [Google Scholar]
- Thuneberg L. Interstitial cells of Cajal: intestinal pacemaker cells? Adv Anat Embryol Cell Biol. 1982;71:1–130. [PubMed] [Google Scholar]
- Tomita T, Pang Y-W, Ogino K. The effects of nickel and cobalt ions on the spontaneous electrical activity, slow waves, in the circular muscle of the guinea-pig gastric antrum. J Smooth Muscle Res. 1998;34:89–100. doi: 10.1540/jsmr.34.89. [DOI] [PubMed] [Google Scholar]
- Tsugeno M, Huang S-M, Pang Y-W, Chowdhury JU, Tomita T. Effects of phosphodiesterase inhibitors on spontaneous electrical activity (slow wave) in the guinea-pig gastric muscle. J Physiol. 1995;485:493–502. doi: 10.1113/jphysiol.1995.sp020745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyama Y, Imaizumi Y, Watanabe M. Effects of cyclopiazonic acid, a novel Ca2+-ATPase inhibitor, on contractile responses in skinned ileal smooth muscle. Br J Pharmacol. 1992;106:208–214. doi: 10.1111/j.1476-5381.1992.tb14316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Burns AJ, Torihashi S, Sanders KM. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J Physiol. 1994;480:91–97. doi: 10.1113/jphysiol.1994.sp020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurons. J Physiol. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Paterson WG. Nitric oxide contracts longitudinal smooth muscle of opossum oesophagus via excitation-contraction coupling. J Physiol. 2001;536:133–140. doi: 10.1111/j.1469-7793.2001.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]