

Abstract
We tested the hypothesis that both stretch-activated channels (SACs) and intracellular calcium ([Ca2+]i) are important in the electrical response of single guinea-pig ventricular myocytes to axial stretch. Myocytes were attached to carbon fibre transducers and stretched, sarcomere length increased by approximately 9 %, and there was a prolongation of the action potential duration. Streptomycin, a blocker of SACs, had no effect upon the shortening, [Ca2+]i transients or action potentials of electrically stimulated, unstretched myocytes, at a concentration of 50 μm, but at 40 μm, prevented any stretch-induced increase in action potential duration. Under action potential clamp, stretch elicited a current with a linear current-voltage relationship that was inward at membrane potentials negative to its reversal potential of −30 mV, in 10 of 24 cells tested, and was consistent with the activation of non-specific, cationic SACs. This current was not seen in any stretched cells that were exposed to 40 μm streptomycin. However, exposure of cells to 5 μm BAPTA-AM, in order to reduce [Ca2+]i transients, also abolished stretch-induced prolongation of the action potential. We conclude that both SACs and [Ca2+]i are important in the electrical response of cardiac myocytes to stretch, and propose that stretch-induced changes in electrical activity and [Ca2+]i may be linked by inter-dependent mechanisms.
Stretch is known to modulate the electrical activity of cardiac muscle (see Lab, 1999, for a review). Stretch-induced alteration of the action potential waveform and initiation of extra-systoles has been reported in single cells (Kamkin et al. 2000), multicellular and whole-heart preparations (Franz et al. 1989, 1992). These disturbances in electrical activity of the myocardium have been linked to clinically observed arrhythmias (Lab, 1999). One possible explanation for these effects is the activation of stretch-activated channels (SACs; Guharay & Sachs, 1984). Stretch also influences the mechanical activity of the heart, increasing contractility over the ascending limb of the length-tension relationship. This increase in tension appears to occur in a two-stage process, termed the rapid and slow response, that affects both the responsiveness of the myofilaments to Ca2+ and the supply of Ca2+ to the myofilaments (e.g. Allen & Kurihara, 1982; Kentish & Wrzosek, 1998).
Intracellular Ca2+ ([Ca2+]i) is known to regulate certain ionic membrane currents, for example the L-type Ca2+ current (ICa,L; Lee et al. 1985), the delayed rectifier K+ current (IK; Tohse, 1990), and the Na+-Ca2+ exchange current (INa-Ca; e.g. Janvier & Boyett, 1996), and as a result of the modulation of these Ca2+-dependent mechanisms, the profile of the action potential (e.g. Du Bell et al. 1991). It is therefore possible that stretch might induce changes in Ca2+ handling that in turn lead to important Ca2+-dependent changes in electrical activity (Lab et al. 1984, Tavi et al. 1998; see Calaghan & White, 1999, for review). However, most previous studies investigating the effects of stretch on the membrane currents and action potentials of single cardiac cells have used added [Ca2+]i buffering that may alter such pathways. The purpose of this study, therefore, was to test the hypothesis that both stretch-activated channels and Ca2+-dependent processes modulate the electrical response to axial stretch in single guinea-pig ventricular myocytes.
Methods
Isolation of guinea-pig ventricular myocytes
Guinea-pigs (weight approximately 300 g) were killed by cervical dislocation following stunning; all procedures conformed with the UK Animals (Scientific Procedures) Act 1986. Left ventricular myocytes were isolated using methods based on those of Frampton et al. (1991). Briefly, hearts were removed and mounted on a Langendorff apparatus and perfused with a Hepes-based isolation solution of the following composition (mm): NaCl 130; KCl 5.4; NaH2PO4 0.4; MgCl2.6H2O 1.4; CaCl2 1.8; Hepes 10; glucose 10; taurine 20; creatine 10; with 1 % bovine serum albumin (pH 7.3) to clear blood from the coronary circulation. Perfusion was then continued with Ca2+-free isolation solution (1.8 mm CaCl2 replaced with 0.1 mm EGTA) for 4 min, followed by 5 min perfusion with isolation solution containing 50 μm Ca2+, 1 mg ml−1 collagenase (type II; Worthington Biochemical Corporation, NJ, USA) and 0.1 mg ml−1 protease (type XIV; Sigma Chemical Co.). The left ventricle was then separated from the rest of the heart, minced, and gently shaken at 37 °C in the collagenase-containing isolation solution. Cells were filtered from this solution at 5 min intervals and resuspended in isolation solution containing 1.8 mm Ca2+.
To reduce [Ca2+]i transients, myocytes were incubated in isolation solution containing 5 μm BAPTA-AM (acetoxymethyl ester form of BAPTA, Molecular Probes, Eugene, OR, USA), dissolved in dimethyl sulphoxide, for 15 min at 22-24 °C, before centrifugation at 40 g for 45 s. The supernatant was removed and cells were resuspended in 1.8 mm Ca2+ isolation solution and left for 30 min before use to allow de-esterification of the AM form of BAPTA. As a confirmation that this caused severe buffering of the [Ca2+]i transient, cells so treated did not shorten when stimulated.
Isolated cells were placed in an experimental chamber on the stage of an inverted microscope (Diaphot, Nikon, Japan). Cells were continuously superfused with a Hepes-based physiological solution containing (mm): NaCl 113; KCl 5; MgSO4.7H2O 1; NaH2PO4 1; CH3COONa 20; CaCl2 2; Hepes 5; glucose 10; with insulin 5 units l−1; pH adjusted to 7.3 with NaOH. Aliquots of a 0.1 m streptomycin sulphate stock solution were added to this solution to give the required final concentration. All experiments were performed at 37 °C.
Measurement of cell length and [Ca2+]i
Some unstretched cells were field stimulated by external platinum electrodes at a frequency of 0.5 Hz and cell length and [Ca2+]i were measured simultaneously. A video image of the cell was monitored using an edge detection system (Crescent Electronics, UT, USA) and the change in cell length following stimulation was used as our measure of contractility. Cells were loaded with the Ca2+-sensitive fluorescent indicator fura-2 AM (Molecular Probes, OR, USA) by incubation for 10 min at 22-24 ° C in 1.8 mm Ca2+ isolation solution containing 3-5 μm fura-2 AM. The ratio of fluorescence emitted at 510 nm in response to alternate excitation with light of 340 and 380 nm (340 nm/380 nm ratio) was our index of [Ca2+]i.
Electrophysiology
Action potentials were measured using sharp microelectrodes containing 600 mm KCl (resistance of 30-60 MΩ) connected to an Axopatch 2B amplifier (Axon Instruments, Inc.). The high chloride content of the microelectrodes did not cause ‘blebbing’ of the myocyte sarcolemma. Action potentials were elicited at a stimulation frequency of 0.5 Hz by 2 ms current pulses, at just above threshold amplitude.
Membrane currents were measured using switch voltage clamp with a microelectrode resistance < 30 MΩ, and a switching frequency of 3 kHz. An oscilloscope was used to check the settling characteristics of the microelectrode. To measure ICa,L cells were voltage clamped at −80 mV, Na+ current was inactivated by a 100 ms prepulse to −40 mV, and ICa,L elicited by a 200 ms pulse from −40 mV to voltages between −80 and +60 mV. ICa,L was measured as the difference between inward peak and current level at the end of the 200 ms pulse (Mitchell et al. 1983). The effects of streptomycin on the rapid (IKr) and slow (IKs) components of IK were tested using electrophysiological separation of the currents (Carmeliet, 1992; Heath & Terrar, 1996) at a stimulation frequency of 0.1 Hz. From a holding potential of −50 mV, depolarisation to +40 mV for 600 ms activated both IKr and IKs, repolarisation to −10 mV (for 4 s) revealed IKs as a deactivating tail current, while subsequent repolarisation to −50 mV revealed deactivating IKr.
Action potential clamp (Doerr et al. 1989) requires the recording of an action potential from a cell; this waveform is then used to voltage clamp the same cell. Free action potentials were recorded as described above (using pClamp7 software, Axon Instruments Inc.). The last recorded action potential, from a train of stable action potentials, was used to provide the voltage clamp protocol. Experimental interventions that alter the free action potential profile induce compensation currents under action potential clamp of opposite polarity to ionic currents. Membrane currents were not normalised to cell size because all comparisons between control and interventions were made in the same cell.
Axial stretching of myocytes
Myocytes were axially stretched by attaching carbon fibres of known compliance close to either end of the myocyte (fibres kindly provided by Prof. J.-Y. Le Guennec, University of Tours, France; Le Guennec et al. 1990; White et al. 1993, 1995; Hongo et al. 1996). Carbon fibre transducers were attached to myocytes by contact. A supple fibre (compliance 22 μm μN−1) was used to stretch the cell. Because of the difference in fibre compliance, stimulated myocytes shorten from this end and bend the fibre during systole. This motion could be tracked by the edge detector (see above) and used as a measure of active force. A stiffer fibre (compliance 0.3 μm μN−1) was placed at the opposite end of the cell. Cells were impaled with microelectrodes behind the stiff fibre in order to protect the microelectrode impalement site from damage during stretching (Fig. 1).
Figure 1. Technique to simultaneously stretch a guinea-pig ventricular myocyte and record electrical activity.
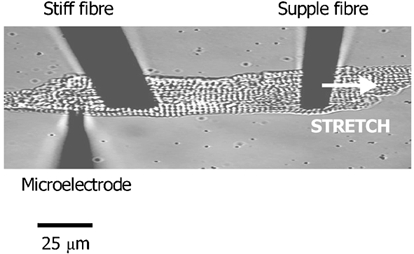
The microelectrode is used to record membrane potential or currents. It is placed behind the stiff fibre to protect the site of impalement during stretch. The supple fibre is used to stretch the cell and its displacement during stimulation is a measure of active force development (increased resting force caused by the stretch cannot be measured in this configuration). Increased sarcomeric spacing is used as the index of stretch.
The index of stretch used in our experiments was an increase in sarcomere length (SL). This was monitored on-line by a Cooley-Tukey fast Fourier transform of the video image of the myocyte, sampled at 50 Hz (Gannier et al. 1993) and calibrated using a 50 × 2 μm graticule. Sarcomere length was not altered by carbon fibre attachment and was stable following increases due to stretch; this was indicative of the absence of damage-induced changes in [Ca2+]i (see White et al. 1993, 1995; Hongo et al. 1996).
Statistical analysis
Data are expressed as means ± s.e.m. of n (number of cells) observations. Statistical significance was set at 0.05 and was tested using Student's paired or unpaired t test, one-way or repeated measures analysis of variance (ANOVA) as appropriate.
Results
Effects of streptomycin in unstretched myocytes
We wished to use 40 μm streptomycin as a pharmacological agent to test for a possible role of SACs in the response to stretch (Gannier et al. 1994). However, in order to ensure the effects of streptomycin were not associated with non-stretch-specific effects upon electrical activity and Ca2+ handling, we initially tested the effect of 50 μm streptomycin in unstretched myocytes. We tested its effect upon cell shortening, [Ca2+]i transients, action potential profile, and upon the major inward (ICa,L) and outward (IKr and IKs) currents of the guinea-pig action potential plateau. We observed no significant effect of 50 μm streptomycin upon either the amplitude or time course of any of these parameters (see Table 1, time course data not given for brevity). These observations strengthened the interpretation of later experiments using 40 μm streptomycin to investigate stretch-activated mechanisms.
Table 1.
Lack of effect of 50μm streptomycin on shortening, [Ca2+]i and electrical activity of unstretched, single guinea-pig ventricular myocytes
| Control | Streptomycin | Cells | |
|---|---|---|---|
| Cell shortening (μm) | 2.82 ± 0.34 | 2.86 ± 0.34 | 12 |
| [Ca2+]i, transient (340 nm/380 nm ratio units) | 0.049 ± 0.005 | 0.049 ± 0.005 | 12 |
| Resting membrane potential (mV) | -82 ± 2 | -81 ± 2 | 6 |
| Action potential duration at 90% repolarisation (ms) | 337 ± 36 | 324 ± 48 | 6 |
| Membrane current amplitude (nA) | |||
| Ica,L | 0.64 ± 0.06 | 0.63 ± 0.05 | 11 |
| IKr | 0.08 ± 0.01 | 0.09 ± 0.01 | 6 |
| IKs | 0.04 ± 0.01 | 0.04 ± 0.01 | 9 |
Mean data for the amplitude of cell shortening and [Ca2+]i, transient, measured simultaneously in fura-2-loaded cells, resting membrane potential and action potential duration at 90% repolarisation, amplitude of L-type Ca2+ current (ICa,L), and the rapid (IKr) and slow (IKs) components of the delayed rectifier K+ current. Exposure to 50 μm streptomycin had no significant effect on any of these parameters (P > 0.05, paired t test).
Effects of axial stretch and streptomycin on the action potentials of myocytes
Axial stretch increased both the amplitude of contraction and the duration of the action potential (APD; Fig. 2). When cells were stretched by approximately 9 %, such that SL increased from 1.81 ± 0.01 to 1.97 ± 0.02 μm, there was a significant (P < 0.05) prolongation of the APD at 20 % (from 91 ± 11 to 114 ± 15 ms), 50 % (from 209 ± 15 to 250 ± 8 ms) and 90 % repolarisation (from 260 ± 14 to 302 ± 16 ms). These effects were seen rapidly upon stretch in 23/26 cells tested; measurements were made within 2 min of stretching. There was no significant (P > 0.05) effect on resting membrane potential (from −78 ± 1 to −78 ± 1 mV) or action potential amplitude (from 126 ± 2 to 127 ± 2 mV).
Figure 2. Effect of axial stretch on the action potential and active force of a guinea-pig ventricular myocyte.

Action potential configuration (A) and active tension (B) before (○) and after (•) a stretch that increased sarcomere length from 1.85 to 2.11 μm. Stretch prolonged the action potential and increased active force.
In contrast, 40 μm streptomycin prevented any significant changes in action potential profile. APD at 20 % (from 159 ± 19 to 179 ± 23 ms), 50 % (from 311 ± 26 to 315 ±25 ms) and 90 % repolarisation (from 353 ± 26 to 357 ± 25 ms), resting membrane potential (from −78 ± 1 to −78 ± 1 mV) and action potential amplitude (from 128 ± 2 to 128 ± 2 mV) were not significantly different when cells were stretched from SL 1.80 ± 0.02 to 1.99 ± 0.01 μm (P > 0.05, n = 8 cells, see Fig. 6).
Figure 6. Comparison of mean effects of stretch on the action potential of guinea-pig ventricular myocytes in the presence and absence of streptomycin or BAPTA.
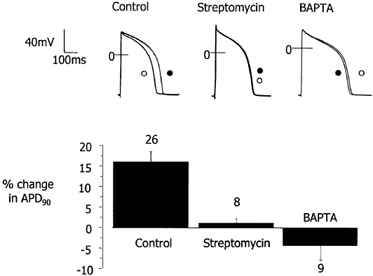
Representative experimental records of action potentials before (○) and after (•) an axial stretch in the presence or absence (Control) of 40 μm streptomycin or the [Ca2+]i buffer BAPTA (upper panel). Mean percentage changes in action potential duration at 90 % repolarisation (APD90; lower panel). In control conditions, stretch prolongs the action potential; this effect is not seen in the presence of 40 μm streptomycin. In the presence of BAPTA there is a tendency for stretch to shorten the action potential. In control cells changes in APD90 were significantly different from zero, streptomycin and BAPTA (P < 0.05, one-way ANOVA and multiple comparison corrected t tests). Numbers refer to the number of cells.
Effect of axial stretch and streptomycin on the membrane currents of myocytes
We next measured the time course and amplitude of currents that might be responsible for the prolongation of the action potential by using action potential clamp. In the absence of stretch and following the rapid decay of capacitive currents, there was little compensation current elicited during the voltage clamp protocol until the cell was stretched (Fig. 3A). The stretch-activated current was calculated as the difference current between the stretched and unstretched conditions (Fig. 3B).
Figure 3. Stretch-activated membrane currents in guinea-pig ventricular myocytes under action potential clamp.
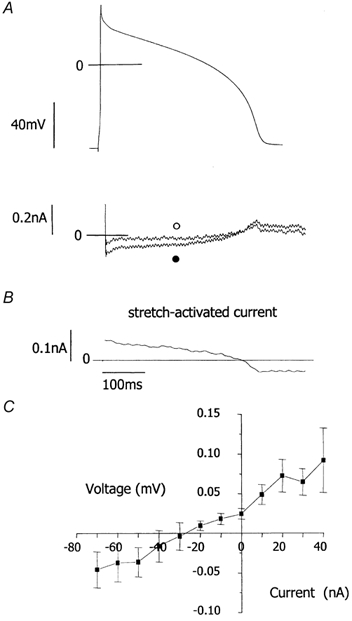
A, upper panel, free action potentials were recorded then used as the waveform to voltage clamp each myocyte. Lower panel, compensation currents (which have a reverse polarity to ionic currents) recorded under action potential clamp before (○) and after (•) a stretch that increased sarcomere length from 1.77 to 2.00 μm. Following decay of large capacitive currents, compensation currents at short sarcomere length are very small. B, stretch-induced current. C, mean current-voltage relationship for the stretch-activated current from 10 cells, having a current of greater than 5 pA at +30 mV. Stretch increased sarcomere length from 1.82 ± 0.01 to 1.98 ± 0.01 μm. The current-voltage relationship was achieved by plotting current (B) against clamp voltage (A, upper panel).
Current-voltage relationships were plotted for the stretch-activated current against membrane potential during the simulated action potential plateau and repolarisation phase (Fig. 3C). In cells stretched from SL 1.82 ± 0.01 to 1.98 ± 0.01 μm, we observed a stretch-activated current with a current amplitude greater than 5 pA at +30 mV, a linear current-voltage relationship with a mean slope conductance of 1.2 nS and a reversal potential close to −30 mV, in 10 cells out of 24 tested.
In contrast stretch-activated currents were not observed in any of the eight cells stretched (from 1.80 ± 0.01 to 1.96 ± 0.04 μm) in the presence of 40 μm streptomycin (Fig. 4A and B). This difference in the probability of observing stretch-activated current (0.42, control versus 0, streptomycin) was significant (P < 0.05, χ2).
Figure 4. Effect of 40 μm streptomycin on stretch-activated currents in guinea-pig ventricular myocytes under action potential clamp.
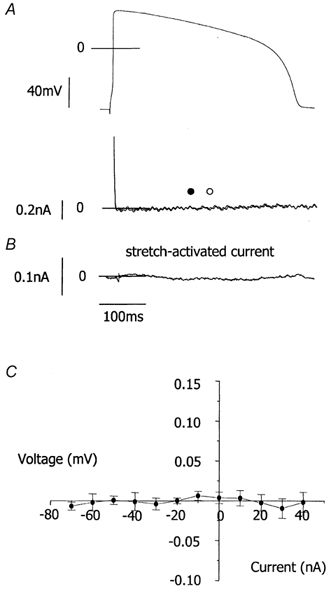
A, upper panel, voltage clamp waveform. Lower panel, compensation currents (which have a reverse polarity to ionic currents) recorded under action potential clamp before (○) and after (•) a stretch that increased sarcomere length from 1.80 to 1.95 μm in the presence of 40 μm streptomycin. B, stretch-induced current. C, mean current-voltage relationship for the stretch-activated current from 8 cells. Stretch increased sarcomere length from 1.80 ± 0.01 to 1.96 ± 0.04 μm. The current-voltage relationship was achieved as in Fig. 3. Stretch-activated currents were not seen in the presence of streptomycin.
Although both the effect of stretch on free APD and stretch-activated currents under action potential clamp were prevented by streptomycin, the current-voltage relationship of this current and its heterogeneous observation suggested it could not fully account for the consistent stretch-induced prolongation of the free APD we observed (see Discussion).
Role of [Ca2+]i in the response of myocytes to axial stretch
We therefore tested for the involvement of Ca2+-dependent mechanisms in the response to stretch. A major Ca2+-regulating pathway is the influx of Ca2+ via ICa,L; however, when cells were voltage clamped and stretched from SL 1.83 ± 0.02 to 2.01 ± 0.01 μm we observed no effect on the amplitude of ICa,L at voltages between −80 and + 60 mV (P > 0.05, n = 12 cells, Fig. 5). Consistent with our experiments under action potential clamp, stretch in conventional voltage clamp led to an inward shift in holding current at −80 mV; in approximately half the cells, the current amplitude varied from a few to 200 pA.
Figure 5. Effect of axial stretch on L-type Ca2+ current in guinea-pig cardiac myocytes.
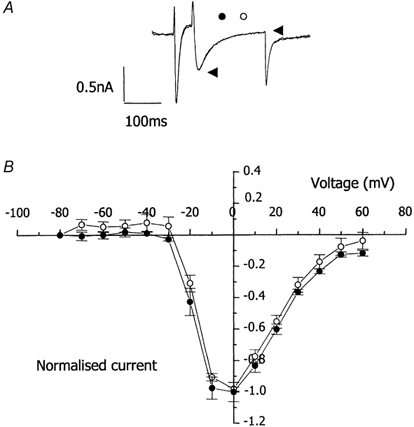
A, membrane currents in a single myocyte voltage clamped at −80 mV, step depolarised to −40 mV for 100 ms to inactivate Na+ currents, then further depolarised to 0 mV for 200 ms to activate L-type Ca2+ current (ICa,L). Amplitude of ICa,L measured as the difference between peak inward current (left arrowhead) and current at the end of the pulse (right arrowhead). Records show superimposed traces of membrane current before (○) and after (•) a stretch that increased sarcomere length from 1.77 to 1.90 μm. B, mean current-voltage relationship for ICa,L from 12 cells before (○) and after (•) stretch that increased sarcomere length from 1.83 ± 0.02 to 2.01 ± 0.01 μm. Current was normalised to maximum pre-stretch ICa,L (0.94 ± 0.11 nA). Stretch did not modify ICa,L.
Another major component of Ca2+ homeostasis in ventricular myocytes is the [Ca2+]i transient. These were greatly reduced by exposure to 5 μm BAPTA-AM. In contrast to cells without added Ca2+ buffering, stretching from SL 1.91 ± 0.01 to 2.05 ± 0.03 μm prevented any significant changes in action potential profile. APD at 20 % (from 174 ± 29 to 166 ± 29 ms), 50 % (from 315 ± 43 to 306 ± 44 ms) and 90 % repolarisation (from 355 ± 43 to 345 ± 44 ms), resting membrane potential (from −77 ± 1 to −77 ± 1 mV) and action potential amplitude (from 120 ± 1 to 120 ± 1 mV) were all unchanged (P > 0.05, n = 12 cells; Fig. 6).
When the change in APD at 90 % repolarisation is compared between treatments it can been seen that the lengthening of the action potential by stretch in control conditions is statistically different from the lack of response in the presence of 40 μm streptomycin and the (non-significant) tendency to shorten in the presence of added Ca2+ buffer (BAPTA; Fig. 6).
Discussion
Streptomycin has been reported to block SACs (see Hamill & McBride, 1996; Pascarel et al. 1997a, for reviews). In multicellular cardiac preparations it has been reported to block stretch-activated changes in electrical activity (e.g. Salmon et al. 1997; Eckardt et al. 2000; Babuty & Lab, 2001), and stretch-induced increases in [Ca2+]i in single myocytes (Gannier et al. 1994). In addition streptomycin can block [Ca2+]i transients and contraction in unstretched preparations (e.g. Belus & White, 2001). However, we observed that 50 μm streptomycin had no effect upon the electrical or mechanical activity of unstretched myocytes nor upon [Ca2+]i transients, whilst 40 μm streptomycin blocked membrane currents and changes in action potential waveform specifically activated by axial stretch. These are important observations, given that we were trying to distinguish between Ca2+- and SAC-dependent mechanisms. In addition, they suggest that of the agents currently readily available to block SACs, streptomycin is the agent of choice (see Pascarel et al. 1997b and Suchyna et al. 2000 for effects of raw tarantula venom compared to isolated GsMTx-4 toxin, and Caldwell et al. 1998 for a discussion of Gd3+).
It is well established that stretch can influence the electrical activity of cardiac muscle (e.g. Lab, 1999). In searching for cellular mechanisms of action, work has concentrated upon establishing a role for SACs. Whole-cell currents consistent with the activation of non-selective cationic SACs have been reported in adult ventricular myocytes by several groups (Sasaki et al. 1992; Kamkin et al. 2000; Zeng et al. 2000; Lei et al. 2001). We too report such a SAC with a current-voltage curve and current amplitude similar to those previously reported (i.e. these studies typically report linear current-voltage curves with reversal potentials between −30 and 0 mV and current amplitudes of a few hundred picoamperes at membrane potentials of approximately ±50 mV).
However, it is equally well established that stretch influences [Ca2+]i by the rapid modulation of myofilament Ca2+ sensitivity and a slower increase in the [Ca2+]i transient (Allen & Kurihara, 1982; Kentish & Wrzosek, 1998; see Calaghan & White, 1999, for review). Many electrogenic processes in the heart are sensitive to Ca2+ (e.g. the inactivation of ICa,L, Lee et al. 1985; the activation of IK, Toshe, 1990, and INa-Ca, Janvier & Boyett, 1996); thus it is possible that stretch could modulate the electrical activity of cardiac muscle, via Ca2+-dependent ion channels, even in the absence of specific SACs.
We observed a consistent stretch-induced prolongation of the APD in control cells, but the APD of some control cells loaded with carbon fibres tended to be shorter than those of other groups. This may be due to chance, reflecting the inherent variability of the APD, but might also be related to the observation by Bett & Sachs (2000a,b) and Lei et al. (2002) that membrane compression in myocytes (in our case, caused by fibre attachment) may activate certain mechanosensitive currents and that the stimulus and effects of compression may differ from axial stretch.
Previous experimental studies have reported that stretch in single guinea-pig ventricular myocytes can produce action potential shortening (White et al. 1993; Lei et al. 2001) or a cross-over effect where the early plateau is depressed and the late repolarisation delayed (Kamkin et al. 2000). However, the present study reports a lengthening of the action potential at all stages of the plateau and repolarisation. Differences in experimental procedures may be the cause of these different observations. In fact there seems to be a relationship between the effects of stretch on the APD and the level of additional Ca2+ buffering (Lei et al. 2001, 37 °C, 5 mm EGTA, APD shortening; Kamkin et al. 2000, 37 °C, 10 μm EGTA, APD cross-over; the present study, 37 °C, no added buffering, APD lengthening). In whole-cell patch clamp studies (Kamkin et al. 2000; Lei et al. 2001) dilution of the intracellular contents by the patch pipette solution might influence some aspects of stretch-activated signalling. The study of White et al. (1993) was performed with microelectrodes, but at 20-25 °C using more flexible carbon fibre transducers, that allowed significant systolic shortening even in unstretched cells (see White et al. 1995). If several mechanisms are altered by stretch it is likely that they will have different activating pathways and/or Q10 values. Thus changes in temperature and intracellular milieu may differentially influence mechanisms and outcomes.
Modelling studies have also predicted stretch-induced APD shortening, cross-over or lengthening in guinea-pig ventricular myocytes. Depending on the characteristics of the SACs, their activation in isolation has been predicted to shorten the guinea-pig ventricular action potential (Sachs, 1994; Kohl et al. 1998), or cause a ‘cross-over’ (Kohl et al. 1998; Reimer et al. 1998). Han et al. (2001) recently suggested that activation of SACs might shorten the APD, primarily through modulation of [Na+]i. However, Kohl et al. (1998) predicted that if increased myofilament Ca2+ sensitivity were the dominant effect of stretch, the associated modulation of [Ca2+]i would prolong APD while Janvier & Boyett (1996) showed increased systolic shortening should prolong APD via increased INa-Ca (as predicted by Lab et al. 1984).
Our observation of stretch-induced changes in APD and of a stretch-induced current with a linear current-voltage relationship and a reversal potential of −30 mV, that was sensitive to 40 μm streptomycin, is strong evidence in support of a role for non-specific cationic SACs in the stretch-induced modulation of cardiac electrical activity. However, the stretch-activated current was not elicited from all stretched cells; this may reflect activation of currents at less than our imposed threshold level, due to prior activation by probe attachment, masking of some outward SAC current by inward Ca2+-activated currents (see below), insufficient stimulus (membrane stress) or channel inactivation at the time of measurement. In contrast to our varied observation of SACs, the stretch-induced prolongation of the free APD was consistently observed. In addition, although the current-voltage relationship of the SACs predicts lengthening of APD at 90 % repolarisation, it predicts shortening at 20 % repolarisation; thus it seems SACs cannot fully explain the effect of stretch on the free action potential and additional mechanisms are involved.
Axial stretch did not increase ICa,L, the major pathway for the generation of depolarising current dependent upon Ca2+ (consistent with previous reports under different experimental conditions; White, 1995; Hongo et al. 1996; Kamkin et al. 2000). However, the observation that the effect of stretch on the APD was abolished by buffering of [Ca2+]i with BAPTA strongly supports the hypothesis that Ca2+ modulation plays a major role in the electrical response of cardiac muscle to stretch.
Because streptomycin and BAPTA were able to abolish the effects of stretch on the free APD in isolation, electrical activity caused by SACs (streptomycin sensitive) and by alterations in Ca2+ handling (BAPTA sensitive) may be interdependent. Because stretch-induced currents elicited under action potential clamp could not fully explain the prolongation of the free action potential, it seems a different balance of mechanisms is operating in the free action potential and action potential clamp states. A possible explanation of our observations is given in Fig. 7. Stretch may cause activation of SACs, initiating prolongation of APD at 90 % repolarisation and Ca2+ entry through SACs (Sigurdson et al.1992; Kim, 1993). In addition stretch will increase diastolic myofilament Ca2+ sensitivity but cause greater systolic shortening (see Fig. 2) and thus greater shortening-induced changes in Ca2+ sensitivity and liberation of Ca2+ from the myofilaments (see Lab et al. 1984; Janvier & Boyett, 1996). This additional Ca2+ may generate depolarising (Ca2+-extruding) INa-Ca. In the free action potential state, an important interaction between voltage-dependent Ca2+ regulation (e.g. INa-Ca) and Ca2+-dependent ionic currents (e.g. ICa,L, IK) results in a new steady state with a general prolongation of the action potential. However, in the action potential clamp state, the clamping of membrane potential will maintain initial Ca2+ extrusion and prevent the development of Ca2+-dependent depolarising currents at plateau potentials, in effect, blocking the pathway at initiation.
Figure 7. Possible interactions between stretch-induced changes in Ca2+ handling and electrical activity in ventricular myocytes.
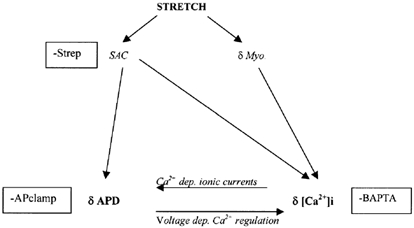
Upon stretch, activation of stretch-activated channels (SACs) and changes in diastolic and systolic myofilament Ca2+ sensitivity (δMyo.) lead to alterations in action potential configuration (δAPD) and intracellular Ca2+ (δ[Ca2+]i). There follows interaction between changes in electrical activity and [Ca2+]i because of the voltage dependence of some calcium-regulating mechanisms (e.g. the Na+-Ca2+ exchanger) and the Ca2+ dependence of certain ionic membrane currents (e.g. L-type calcium current, delayed rectifier K+ current and Na+-Ca2+ exchanger current). This pathway would be interrupted (-) by streptomycin by its action on SACs, by BAPTA via its action on [Ca2+]i and by action potential clamp (APclamp) via its stabilisation of action potential configuration.
If stretch is prolonged, a slow increase in the size of the [Ca2+]i transient, possibly mediated by nitric oxide- (Vila Petroff et al. 2001), endothelin-1- (e.g. Alvarez et al. 1999; Calaghan & White, 2001), angiotensin II- (AT II, e.g. Perez et al. 2001) and/or cAMP-dependent pathways (e.g. Calaghan et al. 1999) will begin to influence this pathway.
In summary, our study is the first to examine the effects of axial stretch on the action potential of single guinea-pig ventricular myocytes at 37 °C in the absence of intracellular dialysis or additional buffering of [Ca2+]i. We provide evidence that alterations in action potential configuration upon axial stretch are associated with the activation of both non-specific cationic SACs and Ca2+-dependent mechanisms.
Acknowledgments
The authors thank Dr S. C. Calaghan for comments on the manuscript. This work was supported by the British Heart Foundation.
References
- Allen D, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez B, Perez N, Ennis I, Camilion de Hurtagon M, Cingolani H. Mechanisms underlying the increase in Ca2+ transient that follow stretch of cardiac muscle: possible explanation of the Anrep effect. Circ Res. 1999;85:716–722. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- Babuty D, Lab M. Heterogeneous changes of monophasic action potential induced by sustained stretch in atrium. J Cardiovasc Electrophysiol. 2001;12:323–329. doi: 10.1046/j.1540-8167.2001.00323.x. [DOI] [PubMed] [Google Scholar]
- Belus A, White E. Effects of antibiotics on the contraction and Ca2+ transients of rat cardiac myocytes. Eur J Pharmacol. 2001;412:121–126. doi: 10.1016/s0014-2999(01)00717-8. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Sachs F. Activation and inactivation of mechanosensistive currents in the chick heart. J Membr Biol. 2000a;173:237–254. doi: 10.1007/s002320001023. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Sachs F. Whole-cell mechanosensitive currents in rat ventricular myocytes activated by direct stimulation. J Membr Biol. 2000b;173:255–263. doi: 10.1007/s002320001024. [DOI] [PubMed] [Google Scholar]
- Calaghan S, White E. The role of calcium in the response of cardiac muscle to stretch. Prog Biophys Mol Biol. 1999;71:59–90. doi: 10.1016/s0079-6107(98)00037-6. [DOI] [PubMed] [Google Scholar]
- Calaghan S, Colyer J, White E. Cyclic AMP but not phosphorylation of phsopholamban contributes to the slow inotropic response to stretch in ferret papillary muscle. Pflügers Arch. 1999;347:780–782. doi: 10.1007/s004240050846. [DOI] [PubMed] [Google Scholar]
- Calaghan S, White E. Contribution of angiotensin II, endothelin-1 and endothelium to the slow inotropic response to stretch in ferret papillary muscle. Pflügers Arch. 2001;441:514–520. doi: 10.1007/s004240000458. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Voltage and time dependent block of the delayed rectifier K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Therap. 1992;262:809–817. [PubMed] [Google Scholar]
- Doerr T, Denger A, Trautwein W. Calcium currents in single SA nodal cells of the rabbit heart studied with action potential clamp. Pflügers Arch. 1989;413:599–603. doi: 10.1007/BF00581808. [DOI] [PubMed] [Google Scholar]
- Du Bell WH, Boyett MR, Spurgeon HA, Talo A, Stern MD, LakattA EG. The cytosolic calcium transient modulates the action potential of rat ventricular myocytes. J Physiol. 1991;436:347–369. doi: 10.1113/jphysiol.1991.sp018554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt L, Kirchof P, Monnig G, Breithardt G, Borggrefe M, Haverkamp W. Modification of stretch-induced shortening of repolarization by streptomycin in the isolated rabbit heart. J Cardiovasc Pharmacol. 2000;36:711–721. doi: 10.1097/00005344-200012000-00005. [DOI] [PubMed] [Google Scholar]
- Frampton JE, Orchard CH, Boyett MR. Diastolic, systolic and sarcoplasmic reticulum [Ca2+] during inotropic interventions in isolated rat myocytes. J Physiol. 1991;437:351–375. doi: 10.1113/jphysiol.1991.sp018600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz M, Burkhoff D, Yue D, SagawA K. Mechanically induced action potential changes and arrhythmia in isolated and in situ canine hearts. Cardiovasc Res. 1989;23:213–223. doi: 10.1093/cvr/23.3.213. [DOI] [PubMed] [Google Scholar]
- Franz M, Cima R, Wang D, Profitt D, Kurtz R. Electrophysiological effects of myocardial stretch and mechanical determinants of stretch-activated arrhythmias. Circulation. 1992;86:968–978. doi: 10.1161/01.cir.86.3.968. [DOI] [PubMed] [Google Scholar]
- Gannier F, Bernengo JC, Jacquemond V, Garnier D. Measurements of sarcomere dynamics simultaneously with auxotonic force in isolated cardiac cells. IEEE Trans Biomed Eng. 1993;12:1226–32. doi: 10.1109/10.250578. [DOI] [PubMed] [Google Scholar]
- Gannier F, White E, Lacampagne A, Garnier D, Le Guennec J-Y. Streptomycin reverses a large stretch-induced increase in [Ca2+]i in isolated guinea-pig ventricular myocytes. Cardiovasc Res. 1994;28:1193–1198. doi: 10.1093/cvr/28.8.1193. [DOI] [PubMed] [Google Scholar]
- Gurahay F, Sachs F. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J Physiol. 1984;352:685–701. doi: 10.1113/jphysiol.1984.sp015317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, McBride DW. The pharmacology of mechanogated membrane ion channels. Pharmacol Rev. 1996;48:231–252. [PubMed] [Google Scholar]
- Han C, Tavi P, Weckstrom M. Modulation of action potential by [Ca2+]i in modelled rat atrial and guinea pig ventricular myocytes. Am J Physiol. 2001;282:H1047, H1054. doi: 10.1152/ajpheart.00573.2001. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Separation of the components of the delayed rectifier potassium K current using selective blockers of Ikr and Iks in guinea-pig ventricular myocytes. Exp Physiol. 1996;81:587–603. doi: 10.1113/expphysiol.1996.sp003961. [DOI] [PubMed] [Google Scholar]
- Hongo K, White E, Le Guennec J-Y, Orchard CH. Changes in [Ca2+]i, [Na+]i and Ca2+ current in isolated rat ventricular myocytes following an increase in cell length. J Physiol. 1996;491:609–619. doi: 10.1113/jphysiol.1996.sp021243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janvier NC, Boyett MR. The role of Na-Ca exchange current in the cardiac action potential. Cardiovasc Res. 1996;32:69–84. [PubMed] [Google Scholar]
- Kamkin A, Kiseleva I, Isenberg G. Stretch-activated currents in ventricular myocytes: amplitudes and arrhythmogenesis effect increase with hypertrophy. Cardiovasc Res. 2000;48:409–420. doi: 10.1016/s0008-6363(00)00208-x. [DOI] [PubMed] [Google Scholar]
- Kentish JC, Wrzosek A. Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol. 1998;506:431–444. doi: 10.1111/j.1469-7793.1998.431bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. Novel cation-selective mechanosensitive ion channel in atrial cell membrane. Circ Res. 1993;72:225–231. doi: 10.1161/01.res.72.1.225. [DOI] [PubMed] [Google Scholar]
- Kohl P, Day K, Noble D. Cellular mechanisms of cardiac mechano-electric feedback in a mathematical model. Can J Cardiol. 1998;14:111–9. [PubMed] [Google Scholar]
- Lab M. Mechanosensitivity as an integrative system in the heart: an audit. Prog Biophys Mol Biol. 1999;71:7–27. doi: 10.1016/s0079-6107(98)00035-2. [DOI] [PubMed] [Google Scholar]
- Lab M, Allen D, Orchard CH. The effectof shortening on myoplasmic calcium concentration and on action potential in mammalian ventricular muscle. Circ Res. 1984;55:825–829. doi: 10.1161/01.res.55.6.825. [DOI] [PubMed] [Google Scholar]
- Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guennec J-Y, Peineau N, Argibay J, Mongo KG, Garnier D. A new method of attachment of isolated mammalian ventricular myocytes for tension recording: length dependence of passive and active tension. J Mol Cell Cardiol. 1990;22:1083–93. doi: 10.1016/0022-2828(90)90072-a. [DOI] [PubMed] [Google Scholar]
- Lei M, Camelliti P, Cooper P, Linz K, Kohl P. Stretch-activated whole-cell currents during the action potential of guinea pig ventricular myocytes. J Physiol. 2001;533.P:40P. [Google Scholar]
- Lei M, Cooper P, Kohl P. Variability in mechanically induced changes in cardiac action potentials: effects of probe attachment. Pflugers Arch. 2002;443:S347–348. abstract. [Google Scholar]
- Mitchell M, Powell T, Terrar D, Twist V. Characteristics of the second inward current in cells isolated from rat ventricular muscle. Proc Roy Soc B. 1983;219:447–469. doi: 10.1098/rspb.1983.0084. [DOI] [PubMed] [Google Scholar]
- Pascarel C, Cazorla O, Brette F, Le Guennec J-Y. Can a specific blocker of mechano-sensitive channels be found ? Curr Top Pharmacol. 1997a;3:229–238. [Google Scholar]
- Pérez N, Cailion de Hurtado M, Cingolani H. Reverse mode of the Na+-Ca2+ exchange after myocardial stretch underlying the mechanism of the slow response. Circ Res. 2001;88:376–382. doi: 10.1161/01.res.88.4.376. [DOI] [PubMed] [Google Scholar]
- Reimer TL, Sobie EA, Tung L. Stretch-induced changes in arrhythmogenesis and excitability in experimentally based heart cell models. Am J Physiol. 1998;44:H431, H442. doi: 10.1152/ajpheart.1998.275.2.H431. [DOI] [PubMed] [Google Scholar]
- Sachs F. Modelling mechanical-electrical transduction in the heart. In: Mow VC, Guliak F, Tran-Son-Tray R, Hochmuth RM, editors. Cell Mechanics and Cellular Engineering. New York: Springer-Verlag; 1994. pp. 308–328. [Google Scholar]
- Salmon AHJ, Mays JL, Dalton GR, Jones JV, Levi AJ. Effect of streptomycin on wall stress induced arrhythmias in the working rat heart. Cardiovasc Res. 1997;34:493–503. doi: 10.1016/s0008-6363(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Mitsuitye T, Noma A. Effects of mechanical stretch on membrane currents of single ventricular myocytes of guinea pig heart. Jap J Physiol. 1992;42:957–970. doi: 10.2170/jjphysiol.42.957. [DOI] [PubMed] [Google Scholar]
- Sigurdson W, Ruknudin A, Sachs F. Calcium imaging of mechanically induced fluxes in tissue-cultured chick heart: role of stretch-activated ion channels. Am J Physiol. 1992;262:H1110, H1115. doi: 10.1152/ajpheart.1992.262.4.H1110. [DOI] [PubMed] [Google Scholar]
- Suchyna T, Jonhson J, Hamer K, Leykam J, Gage D, Clemo H, Baumgarten C, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch activated channels. J Gen Physiol. 2000;115:583–598. doi: 10.1085/jgp.115.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavi P, Han C, Weckstrom M. Mechanisms of stretch-induced changes in [Ca2+]i in rat atrial myocytes: role of increased troponin C affinity and stretch-activated ion channels. Circ Res. 1998;83:1165–1177. doi: 10.1161/01.res.83.11.1165. [DOI] [PubMed] [Google Scholar]
- Tohse N. Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am J Physiol. 1990;258:H1200, H1207. doi: 10.1152/ajpheart.1990.258.4.H1200. [DOI] [PubMed] [Google Scholar]
- Vilaz Petroff M, Kim S, Pepe S, Dessy C, Marbán E, Balligand J-L, Sollot S. Endogenous nitric oxide mechanisms mediate stretch-dependency of Ca2+-release in cardiomyocytes. Nature Cell Biol. 2001;3:867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- White E. The lack of effect of increasing cell length on L-type calcium current in isolated ferret ventricular myocytes. J Physiol. 1995;483.P:13P. abstract. [Google Scholar]
- White E, Boyett MR, Orchard CH. The effect of mechanical loading and changes on single guinea pig ventricular myocytes. J Physiol. 1995;481:93–107. doi: 10.1113/jphysiol.1995.sp020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Le Guennec J-Y, Nigretto JM, Gannier F, Argibay J, Garnier D. The effect of increasing length on auxotonic contractions; membrane potential and intracellular calcium transients in single guinea pig ventricular myocytes. Exp Physiol. 1993;78:65–78. doi: 10.1113/expphysiol.1993.sp003671. [DOI] [PubMed] [Google Scholar]
- Zeng T, Bett G, Sachs F. Stretch-activated whole cell currents in adult rat cardiac myocytes. Am J Physiol. 2000;278:H548, H557. doi: 10.1152/ajpheart.2000.278.2.H548. [DOI] [PubMed] [Google Scholar]