

Abstract
We studied quinidine block of Kv1.4ΔN, a K+ channel lacking N-type inactivation, expressed in Xenopus ooctyes. Initially, quinidine intracellularly blocked the open channel so rapidly it overlapped with activation. This rapid open channel block was reduced (non-additively) by interventions that slow C-type inactivation: [K+]o elevation and an extracellular lysine to tyrosine mutation (K532Y). These manipulations reduced the affinity of rapid open channel block ≈10-fold, but left the effective electrical distance unchanged at ≈0.15. Following rapid open channel block, there were time-dependent quinidine effects: the rate of inactivation during a single depolarisation was increased, and repetitive pulsing showed use dependence. The rate of recovery from the time-dependent aspect of quinidine block was similar to recovery from normal C-type inactivation. Manipulations that prevented the channel from entering the C-type inactivated state (i.e. high [K+]o or the K532Y mutation) prevented the development of the time-dependent quinidine-induced inactivation. The concentration dependence of the rapid block and the time-dependent quinidine-induced inactivation were similar, but the time-dependent component was strongly voltage sensitive, with an effective electrical distance of 2. Clearly, this cannot reflect the permeation of quinidine through the electric field, but must be the result of some other voltage-sensitive change in the channel. We propose that quinidine promotes the entry of the channel into a C-type inactivated state in a time- and voltage-dependent manner. We developed a mathematical model based on these results to test the hypothesis that, following rapid open channel block, quinidine promotes development of the C-type inactivated state through a voltage-dependent conformational change.
Voltage-dependent potassium currents play an essential role in repolarisation and pacemaker activity in the heart. In response to a depolarising step in potential, voltage-gated potassium channels undergo activation, a transition through which the channel enters an open, or conducting, conformation (Hille, 2001). Following activation, most voltage-gated potassium channels transition to a stable non-conducting state, through a process called inactivation. The rate at which native channels recover from inactivation is an important determinant of their role during repolarisation, since channels that have not recovered by the time a subsequent stimulus arrives will not be able to open (Hodgkin & Huxley, 1952; Rasmusson et al. 1998; Hille, 2001).
Early studies by Aldrich and co-workers clearly established that N-type inactivation in Shaker K+ channels results from block of the inner pore of the open channel by the N-terminal region (Zagotta et al. 1990). At about the same time, another form of inactivation was identified in many K+ channels: C-type inactivation (Hoshi et al. 1990, 1991). Although C-type inactivation was initially identified in the absence of N-type inactivation, both N- and C-type inactivation co-exist in some K+ channels, including Kv1.4 (Hoshi et al. 1991; Rasmusson et al. 1995a). The slow inactivation found in Na+ and Ca2+ channels results from a mechanism that resembles K+ channel C-type inactivation (Zhang et al. 1994; Balser et al. 1996; Wang & Wang, 1997; Todt et al. 1999). Therefore, C-type inactivation may be a general gating mechanism that underlies channel inactivation for many different cation channel types.
C-type inactivation can be slowed by K+ or TEA+, which bind to sites on the extracellular side of the protein (Choi et al. 1991; Lopez-Barneo et al. 1993). Particular residues on the extracellular side of the S6 transmembrane segment (Hoshi et al. 1990) and at the C-terminal end of the H5 loop can markedly affect the rate of development of C-type inactivation (Lopez-Barneo et al. 1993). These initial observations led to the view that C-type inactivation involved a small-scale conformational change near the external mouth of the pore. This external localisation of C-type inactivation was further supported by the finding that C-type inactivation changed the extracellular solute accessibility of a limited number of residues near the outer mouth of the pore in the Shaker K+ channel (Liu et al. 1996). However, recent studies have demonstrated that residues at the C-terminal end of the S6 transmembrane segment can also modulate the rate of C-type inactivation (Li et al. 2003), indicating that C-type inactivation may be more sensitive to overall channel conformation than originally thought.
C-type inactivation in both Shaker and mammalian K+ channels is accelerated when the intracellular region of the channel pore is blocked by either the N-terminal domain or open channel blockers (Rasmusson et al. 1995a,b, 1998; Baukrowitz & Yellen, 1995, 1996a,b; Holmgren et al. 1997; Li et al. 2003). In a similar way, interventions that slow C-type inactivation in the HERG (human ether à go go-related gene) channel also reduce the binding affinity of the intracellularly acting methanesulfonanilide drug E-4031 (Wang et al. 1997). These data suggested that there may be a link between the binding of drugs to the intracellular side of the channel and the conformational transition involved in C-type inactivation (Wang et al. 1997; Ficker et al. 1998).
Quinidine is a commonly used anti-arrhythmic agent which blocks K+ channels, including the cardiac transient outward current, Ito (Imaizumi & Giles, 1987; Balser et al. 1991), and other cloned K+ channels (Yatani et al. 1993; Yang & Roden, 1996; Yang et al. 1997; Yeola & Snyders, 1997). The blocking action of quinidine is state dependent (i.e. quinidine binds preferentially to open channels) in native myocyte currents and wild-type Kv1.5 and Kv1.4 channels (Slawsky & Castle, 1994; Clark et al. 1995; Snyders & Yeola, 1995; Fedida, 1997). Quinidine does not affect the kinetics of activation (Fedida, 1997).
This study examines the role of intracellular quinidine binding in catalysing C-type inactivation. Kv1.4 was studied because its C-type inactivation properties are well characterised (Rasmusson et al. 1995a), and it is thought to contribute to the generation of Ito in the endocardial region of the left ventricle (Wang et al. 1999; McKinnon, 1999; Wickenden et al. 1999; Tseng, 1999; Brahmajothi et al. 1999; Nerbonne, 2000). We used an N-terminal deletion construct of Kv1.4 (Kv1.4ΔN), which lacks rapid N-type inactivation, but exhibits robust C-type inactivation (Comer et al. 1994; Rasmusson et al. 1995a).
We demonstrate that quinidine block of the Kv1.4ΔN channel is a complex process, involving more than one conformational state. Initially, quinidine blocks the open channel with rapid kinetics on the same time scale as activation. Subsequently, the quinidine-bound channel enters a C-type inactivated state in a time- and voltage-dependent manner. Recovery from this state is the same as recovery from C-type inactivation, and governs the use dependence of quinidine block.
Methods
Molecular biology
The constructs and sequences of the cDNAs ferret Kv1.4 (fKv1.4) and its mutants fKv1.4ΔN and fKv1.4[K532Y]ΔN used in this study have been previously described (Comer et al. 1994; Rasmusson et al. 1995a). Ferret Kv1.4 channels possess both fast (N-type) and slow (C-type) inactivation (Comer et al. 1994; Rasmusson et al. 1995a). Removal of residues 2-146 from the N-terminal domain (fKv1.4ΔN) results in the loss of the fast component of inactivation but leaves C-type inactivation. Mutation of the lysine at position 532 to a tyrosine in the fKv1.4ΔN channel (fKv1.4[K532Y]ΔN) results in a marked slowing of the rate of development of C-type inactivation (Rasmusson et al. 1995a).
Oocytes were collected from mature female Xenopus laevis (Xenopus One, Ann Arbor, MI, USA) under anaesthesia (immersion in 1.5 g l−1 tricaine). Frogs were humanely killed after the final collection. All procedures were approved by the Institutional Animal Care and Use Committee of the University at Buffalo, State University of New York and conformed to all federal guidelines and with the principles of the UK Animals (Scientific Procedures) Act 1986. The follicular layer was removed enzymatically by placing the lobes in a collagenase-containing, Ca2+-free OR2 solution (mm: 82.5 NaCl, 2 KCl, 1 MgCl2 and 5 Hepes, pH 7.4, with 1-2 mg ml−1 collagenase (Type I, Sigma). The oocytes were gently shaken for about 2 h and collagenase activity was then halted by bovine albumin as previously described (Comer et al. 1994). Defolliculated oocytes (stage V-VI) were then injected with one of four transcribed cRNAs (up to 50 nl) using a Nanoject microinjection system (Drummond Scientific Co., Broomall, PA, USA) and incubated at 18 °C for 24-72 h in an antibiotic-containing Barth's solution (mm: 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 0.33 Ca(NO3)2, 0.41 CaCl2 and 10 Hepes, pH 7.4, with 2 % (v/v of 100 × stock) antibiotic-antimycotic (Sigma).
Electrophysiology
Oocytes were clamped using a two-microelectrode bath clamp amplifier (OC-750A, Warner Instruments Corp., Hamden, CT, USA) as has been described in detail elsewhere (Comer et al. 1994). Microelectrodes were fabricated from 1.5 mm o.d. borosilicate glass tubing (TW150F-4, WPI) using a two-stage puller (L/M-3 P-A, Adams & List Associates Ltd, Great Neck, NY, USA) to produce electrodes with resistances of 0.6-1.5 MΩ when filled with 3 m KCl. During recording, oocytes were continuously perfused with control solution (mm: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH) or 98K solution (mm: 98 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH). Whenever quinidine was used, 10 min of perfusion time was used to allow equilibration of quinidine with the oocyte. Unless otherwise stated, after this wash-on period, a series of 500 ms depolarising pulses (from −90 to +50 mV at a frequency of 1 Hz for 1 min) was employed to ensure steady-state block before beginning experimental protocols. Currents were recorded at room temperature (21-23 °C) and were filtered at 2.5 kHz. In all experiments except those using the cut-open oocyte clamp technique, quinidine was applied to the extracellular bath solution. The cut-open oocyte technique was used as described previously (Bezanilla et al. 1991; Comer et al. 1994).
Data analysis
Data were recorded on videotape using an A/D VCR adaptor (Medical Systems Corporation, Greenvale, NY, USA) and were digitised and analysed directly using pCLAMP 6 software (Axon Instruments, CA, USA), Sigmaplot (Jandel Scientific, CA, USA) or Microsoft Excel (Microsoft, WA, USA). Unless otherwise stated, raw data traces from two-microelectrode voltage-clamp recordings were not leakage or capacitance subtracted. Data from cut-open oocyte experiments were leakage and capacitance subtracted. The pulse protocols used and the equations used to fit the data are given in each figure legend. Data are shown as means ± s.e.m. Confidence levels were calculated using Student's paired t test.
Computer modelling
A mathematical model of channel gating and quinidine block was based on a core model of activation derived from the analysis of previously published activation kinetics (Comer et al. 1994). Briefly, steady-state equations for activation, and time constants for activation and deactivation were developed. Assuming a fourth-order activation process produced a model of the form:
| (1) |
where C and O represent the closed and open states, respectively, and the voltage-dependent rate constants α and β were as follows:
| (2) |
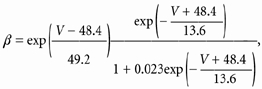 |
(3) |
where V is voltage. All values were calculated by fitting the data as described in Comer et al. (1994).
Additional components of drug block and inactivation were added as described in the text. The equations were solved using a first-order Euler method implemented in an Excel spreadsheet (Microsoft) and in Mathematica (Wolfram Research, IL, USA). All simulations were tested for insensitivity to time step size to ensure accuracy of numerical integration.
Results
Rapid open channel block
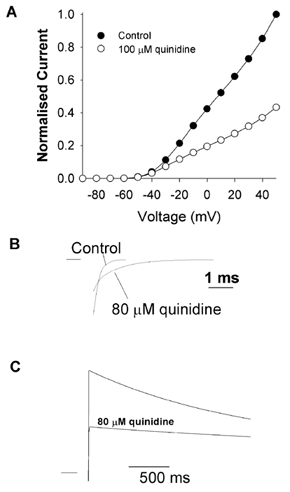
Members of the voltage-gated potassium Kv1 family of channels are blocked when quinidine binds to an intracellular site, which is accessible only when the channel is open (Snyders et al. 1992; Yatani et al. 1993; Yeola et al. 1996; Yang et al. 1997; Fedida, 1997; Chen & Fedida, 1998; Zhang et al. 1998). For quinidine binding to Kv1.4, open channel block is rapid and overlaps with the time course of activation, making it difficult, if not impossible, to resolve kinetically (cf. quinidine block of Kv1.5 channels, Snyders et al. 1992). The initial consequence of the rapid block is a reduction in peak outward current. Figure 1 shows two-electrode voltage-clamp current recordings from an fKv1.4ΔN channel expressed in Xenopus oocytes in the absence and presence of quinidine. Quinidine reduced the magnitude of the peak current, and resulted in an increase in the rate of inactivation during the remainder of the depolarising pulse (control τinactivation was 2.692 ± 0.317 s compared to the dominant τinactivation of 1.805 ± 0.246 s with 100 μm quinidine (n = 6, P < 0.05)). Peak current- voltage relationships from control and quinidine-treated oocyte groups were plotted against clamp potential. The peak current was blocked by 52.2 ± 5 % (n = 3) when depolarised to +50 mV in 100 μm quinidine. There was a weak voltage dependence to the action of quinidine, which is typical of open channel block.
Figure 1. Quinidine blocks fKv1.4ΔN channels.
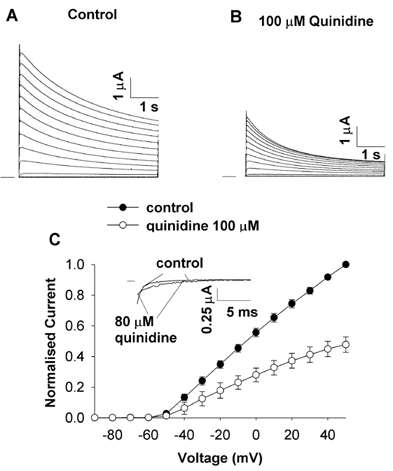
Current recordings from two-electrode voltage clamp of oocytes expressing fKv1.4ΔN. Currents were elicited by a series of 5 s depolarising pulses between −90 and +50 mV in 10 mV increments, applied at 0.03 Hz. A, control currents in 2 mm[K+]o. B, 100 μm quinidine decreased the peak currents at potentials positive to the activation threshold (-50 mV). The rate of inactivation was also increased. C, peak current-voltage relationships in control and 100 μm quinidine. The currents were normalised to the peak current at +50 mV under control conditions. Data are shown as means ± s.e.m., n = 3. Inset, deactivation current tails obtained when repolarising from +50 to −120 mV in control and 80 μm quinidine exhibiting the ‘cross-over’ phenomenon.
Additional evidence for open channel block is found in deactivation, the reverse process to activation. When an open channel is repolarised, there is a ‘tail’ current as the open channels gradually return to the closed state. When an open-but-blocked channel is repolarised, the drug must first dissociate from the binding site before the channel can close. This extra step in response to repolarisation results in a slowing of the time course of the deactivation tail currents. This apparent slowing of deactivation, coupled with a reduction in the initial magnitude of the peak of the tail, results in a ‘cross-over’ when control and drug-bound deactivation tails are compared. This phenomenon has been described in some detail for the actions of quinidine on Kv1.5 channels (Snyders et al. 1992).
We observed a similar phenomenon in the Kv1.4ΔN channels in the cut-open configuration (Fig. 1C, inset). After activating the channel by depolarising from −90 to +50 mV for 10 ms, repolarising back to −120 mV resulted in a slowing of the deactivation tail currents. The time constant of deactivation in control conditions was τdeactivation,control = 4.8 ± 0.9 ms whereas in the presence of 80 μm quinidine it increased to τdeactivation,quinidine = 7.2 ± 1.9 ms (n = 4, P < 0.05). These results are a further indication that the reduction in peak current is due to a rapid open channel block by quinidine. The kinetics of quinidine rapid open channel block are fast so it is effectively ‘instantaneous’ or ‘time independent’.
C-type inactivation in Kv1.4 channels can be inhibited by interventions such as elevating [K+]o and mutating an extracellular lysine to a tyrosine (fKv1.4[K532Y]ΔN, see Fig. 2; Rasmusson et al. 1995a). We examined the effects of these two interventions on quinidine rapid open channel block. In the presence of 2 mm [K+]o, 80 μm quinidine produced a 49 ± 8 % (n = 4) block of the fKv1.4ΔN peak current (Fig. 3). By contrast, in the presence of 98 mm [K+]o, 400 μm quinidine was needed to achieve 47 ± 2 % (n = 3) block of the peak current in the same channel.
Figure 2.
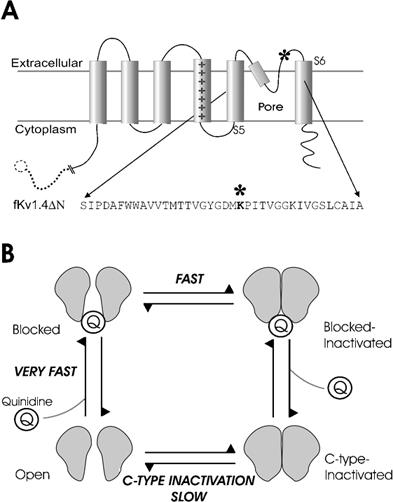
A, schematic representation of one subunit of the fKv1.4ΔN channel. The amino acid sequence of the residues in the pore-forming region and N-terminal end of S6 are shown, with an asterisk marking the approximate location of the pore-mouth lysine to tyrosine mutation at position 532. The dashed line and circle indicate the truncation of the N-terminus. B, the fundamental hypothesis of this paper is that there are two related components of quinidine block: a very fast open channel block and a slowly developing component. The slow component is a C-type inactivation that is catalysed by quinidine. This is illustrated schematically by the cartoon, which shows that the open channel usually inactivates slowly via regular C-type inactivation, but when quinidine is bound it inactivates more rapidly.
Figure 3. Interventions that are known to slow C-type inactivation result in a reduction of quinidine binding affinity for fKv1.4ΔN channels.
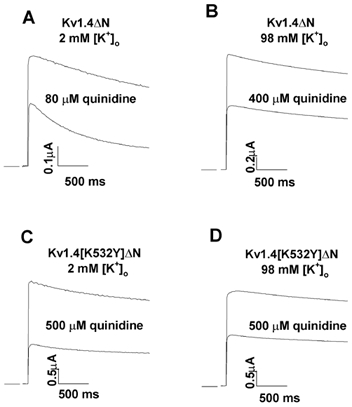
Depolarising pulses from −90 to +50 mV were applied at a frequency of 0.03 Hz. A, representative fKv1.4ΔN currents in 2 mm[K+]o, in control and 80 μm quinidine. Quinidine reduced the peak current by ≈50 %. B, in 98 mm[K+]o, a 5-fold larger quinidine concentration, 400 μm, was required to achieve comparable block of the peak current. C, when 500 μm quinidine was applied to the fKv1.4[K532Y]ΔN mutant with 2 mm[K+]o the peak current was reduced by ≈50 %. D, raising [K+]o to 98 mm did not affect the concentration of quinidine required to reduce peak current by ≈50 % in the mutant channel.
When the fKv1.4[K532Y]ΔN channel was studied in 2 mm [K+]o it took 500 μm quinidine to produce a 52 ± 3 % (n = 3) block of the peak current (rapid open channel block). Raising [K+]o to 98 mm has little effect on C-type inactivation in this mutant (Rasmusson et al. 1995a). Similarly, rapid open channel block of the mutated channel showed no [K+]o dependence.
The concentration dependence of rapid open channel quinidine block of peak fKv1.4ΔN and fKv1.4[K532Y]ΔN current at 2 and 98 mm [K+]o is shown in Fig. 4. The apparent dissociation constant, KD, for rapid open channel block of fKv1.4ΔN in 2 mm [K+]o was 76.4 μm(n = 4), which is comparable to the KD value (63.2 μm) obtained in rat Kv1.4(Δ3-25) (Zhang et al. 1998). The apparent affinity deduced using the two-electrode clamp technique was the same as that obtained by the cut-open oocyte technique (Fig. 4). In the latter experiments, quinidine was applied intracellularly while the external solution was effectively maintained quinidine-free, which confirms that quinidine acts at an intracellular site. The apparent dissociation constant for quinidine was reduced by increasing [K+]o to 98 mm (KD = 374.7 μm, n = 9) or a lysine to tyrosine mutation at position 532 (K532Y; KD = 435.0 μm in 2 mm [K+]o(n = 3) and KD = 484.8 μm in 98 mm [K+]o(n = 6)). These interventions produced a 4.9- to 6.3-fold reduction in the apparent affinity for rapid open channel quinidine binding, which parallels the effects of these interventions on C-type inactivation. The similarity of the responses of quinidine binding and C-type inactivation to these manipulations suggests there may be some common mechanisms involved in quinidine binding and C-type inactivation.
Figure 4. Dose-response relationships for quinidine acting on fKv1.4ΔN and fKv1.4[K532Y]ΔN channels at 2 and 98 mm[K+]o.
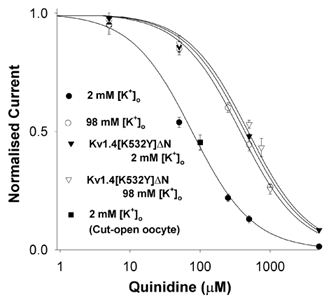
In the fKv1.4ΔN channel, when [K+]o was increased from 2 mm (filled circles) to 98 mm (open circles) the apparent KD at +50 mV for quinidine was increased ≈5-fold. The difference in KD between fKv1.4ΔN and fKv1.4[K532Y]ΔN (filled triangles) with 2 mm[K+]o was 5.7-fold. Increasing [K+]o from 2 to 98 mm (open triangles) in the mutant channel increased the KD by a further 10 %. Data were obtained with the two-electrode voltage-clamp technique. Further data were added from the cut-open oocyte technique (filled square) when 100 μm quinidine was applied intracellularly while the extracellular quinidine concentration was effectively zero. All values shown are normalised to the peak current in the absence of drug in 2 or 98 mm[K+]o. Assuming a 1:1 binding stoichiometry between drug and receptor, curves were derived by fitting the data to the equation: f =KD/(KD+[D]), where f is fractional current, KD is the apparent dissociation constant, and [D] is the quinidine concentration. Symbols and error bars are means ± s.e.m.
The mutual dependence of quinidine binding and C-type inactivation on [K+]o and the K532Y mutation in Kv1.4 channels is in direct contrast to the relationship between E-4031 binding and HERG channel inactivation (Wang et al. 1997). E-4031 binds intracellularly to the HERG channel, with a resulting decrease in current. Double mutation of an extracellular serine and glycine to cysteines removes C-type inactivation in the HERG channel, as does raising [K+]o. However, in contrast to our results with quinidine and the Kv1.4 channel, elevating [K+]o still reduces the affinity of E-4031 even in the C-type inactivation-deleted mutant HERG channel. This indicates that C-type inactivation and E-4031 binding in HERG channels are independent mechanisms (Wang et al. 1997).
Given the dramatic change in quinidine affinity that occurs with manipulations that are known to inhibit C-type inactivation, it was important to determine whether other properties of drug-channel interactions change following quinidine binding. We did this by calculating the ‘fractional electrical distance’ of open channel block (cf. Snyders & Yeola, 1995; see Bett & Rasmusson, 2002 for a review). Rapid open channel block typically involves movement of the drug into the pore region of the channel. In the case of quinidine blocking Kv1.4 channels, the positively charged quinidine must traverse a portion of the transmembrane field, with the result that the block is voltage dependent. We determined the effect of the transmembrane electrical field on the rapid open channel block by plotting the fractional block of fully activated currents (Iquinidine/Icontrol) against the test potential (Fig. 5). The fraction of the electrical field through which the drug passes (δ, fractional electrical distance) was calculated using the Woodhull equation (Woodhull, 1973):
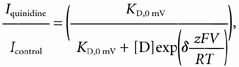 |
(4) |
where z, F, R and T have their usual meanings, KD,0 mV is the binding constant at 0 mV, [D] is the concentration of drug, and V is the membrane voltage.
Figure 5. Quinidine block of fKv1.4ΔN channels is voltage dependent.
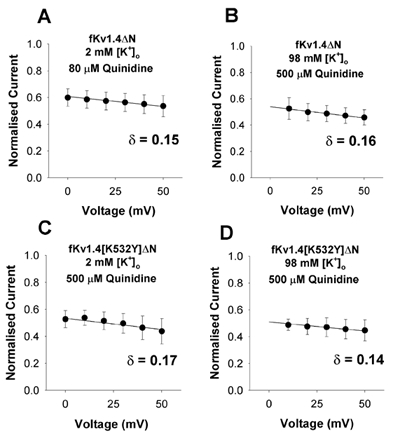
Kv1.4ΔN and Kv1.4[K532Y]ΔN channels were studied in both 2 and 98 mm[K+]o. Normalised current (Iquinidine/Icontrol) is plotted against voltage for positive potentials where steady-state activation is 1. The equation f =KD,0 mV/(KD,0 mV+[D]exp(δZFV/RT)) was fitted to the data, where z, F, R and T have their usual meanings, δ represents the fraction of the transmembrane electrical field sensed by quinidine, and KD,0 mV stands for the apparent dissociation constant at 0 mV. δ was 0.15 ± 0.05, 0.16 ± 0.04, 0.17 ± 0.05 and 0.14 ± 0.05 in A, B, C and D, respectively (n = 3).
The data in Fig. 5 suggest that quinidine block of fKv1.4ΔN channels is weakly voltage dependent, with the drug sensing approximately 16 % of the total electrical field across the membrane from an intracellular site. In addition, the interventions that slowed C-type inactivation and disrupted quinidine binding affinity (raising [K+]o and/or mutating K532Y) had no statistically significant effect on the voltage dependence of rapid open channel quinidine block (n = 3, P > 0.05, between any 2 groups by Student's paired t test). These experiments suggest that despite a dramatic change in affinity, neither increasing [K+]o nor mutating the lysine to tyrosine changes the intracellular site of action or the depth of interaction within the intracellular pore.
Use-dependent block
Rapid open channel block of Kv1 channels by quinidine develops into a secondary time-dependent phenomenon which develops and recovers with relatively slow kinetics. This time-dependent process has many properties in common with conventional C-type inactivation, and gives rise to the use dependence of quinidine block of the Kv1.4 channel. To determine the longer-term effects of exposing the fKv1.4ΔN channel to quinidine we applied a series of 500 ms depolarising pulses from −90 to +50 mV with a frequency of 1 Hz for a period of 1 min. Figure 6 shows the peak currents elicited by this protocol, before and after exposure to 80 μm quinidine, normalised to the first peak current under control conditions.
Figure 6. Frequency-dependent block of fKv1.4ΔN channels by quinidine.
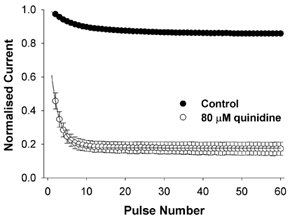
Two-electrode voltage-clamp currents were elicited by applying a series of depolarising pulses from −90 to +50 mV with a frequency of 1 Hz. The peak current was normalised to the maximum control value without drug. In control cells, there was a use-dependent reduction in the magnitude of the peak current. When cells were exposed to 80 μm quinidine for 10 min before stimulation, there was a reduction in the magnitude of the first peak current (compared to the control value) and then a use-dependent component. The use-dependent reduction in current was much greater than that seen in control.
The first pulse of the pulse train in the presence of 80 μm quinidine showed a decrease relative to the pre-drug control, due to rapid open channel block. The magnitude of this reduction in current was similar to that seen under steady-state conditions when a sufficiently long recovery time was allowed between test pulses.
In both control and 80 μm quinidine there was a use-dependent reduction in the peak current when stimulated at 1 Hz, but in 80 μm quinidine the use-dependent decrease was much greater than in control. In control (in 2 mm [K+]o) the peak current decayed mono-exponentially from 100 to 86 ± 1 % (n = 3). By contrast, in 80 μm quinidine, the current decreased from 46 ± 5 % (n = 3; due to initial rapid open channel block) to 17 ± 4 % (n = 3) in the steady state. The use dependence and quinidine-induced increase in inactivation rate was completely abolished by raising [K+]o to 98 mm or by the K532Y mutation and could not be restored by high concentrations of quinidine.
The time-dependent progression of the channel from the rapid open channel block conformation into a quinidine-induced inactivated state was also apparent during a single depolarising step from −90 to +50 mV. In the absence of drug, the rate of inactivation of fKv1.4ΔN was mono-exponential with a time constant of 2.603 ± 0.2 s (n = 3) in control conditions. In the presence of quinidine, inactivation became bi-exponential, with a dominant fast exponential (Fig. 7). Both time constants of inactivation were concentration dependent, with an increase in quinidine concentration resulting in an increase in the rate of inactivation.
Figure 7. Quinidine binding kinetics.
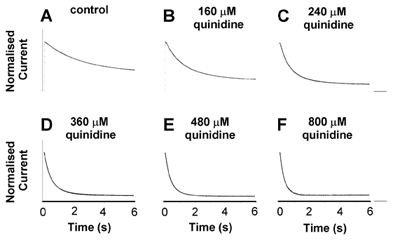
Six second depolarising steps from −90 to +50 mV were applied at a frequency of 0.03 Hz. Recordings were made in 2 mm[K+]o with between 0 and 800 μm quinidine. For comparison, all current traces were normalised to the peak values under control conditions. The smooth continuous line superimposed on each trace is the best fit of an exponential function, used to determine the inactivation time constant(s). The control trace was best fitted by a mono-exponential function (A), whereas in the presence of 160-800 μm quinidine, inactivation was best fitted by a bi-exponential function (B-F).
Use-dependent block is generally voltage sensitive, and dependent on association rates at positive potentials and dissociation/recovery rates at negative potentials. We examined the kinetic behaviour of recovery from block at negative potentials. Previously, quinidine was shown to slow the rate of recovery of Ito in cardiac muscle substantially (Imaizumi & Giles, 1987). We examined the effect of quinidine on the rate of recovery from inactivation in fKv1.4ΔN channels using a standard two-pulse protocol (Fig. 8). The recovery seen in control conditions was similar to that in 80 μm quinidine, although a small statistically significant slowing in the rate of recovery was noted in the presence of the drug. Slow recovery is a characteristic of C-type inactivation in these channels. The slowness of recovery of drug-blocked channels and the similarity between the rate of recovery in control and with quinidine suggests that the recovery process in both cases is the same, i.e. recovery of the blocked-inactivated channel is dominated by the rate of recovery from C-type inactivation. This is consistent with the idea that quinidine promotes the development of C-type inactivation at positive potentials.
Figure 8. Effect of quinidine on the rate of recovery from inactivation in fKv1.4ΔN.
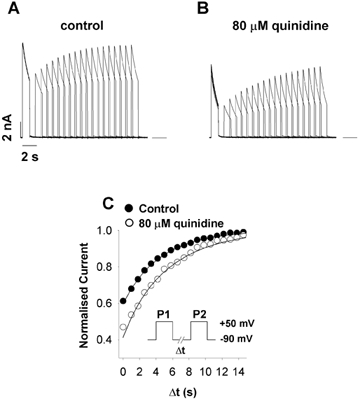
A, fKv1.4ΔN was depolarised from −90 to +50 mV for 1 s (P1) with a variable duration inter-pulse interval (Δt) followed by a pulse (P2) to +50 mV (stimulation rate = 0.03 Hz, [K+]o = 2 mm). B, same protocol as described in A, but with 80 μm quinidine. C, peak currents during P2 were normalised to peak P1 currents and plotted as a function of Δt. The filled and open circles represent control and quinidine-treated data, respectively. Data were fitted using the equation: f = 1 – (Aexp(-t/τ)), where f is fractional current, A is the amplitude of the current, t is duration (in s) and τ is the time constant. Quinidine only slightly slowed the rate of recovery. The mean time constants for recovery were 4.873 ± 0.126 s (n = 5) in control and 5.358 ± 0.086 s (n = 5) in the quinidine-treated group (P < 0.002 by Student's paired t test). This similarity in recovery rate suggests that the ability of channels to conduct on succeeding depolarisations is dominated by recovery from inactivation and not by drug dissociation.
Quinidine promotion of C-type inactivation is illustrated schematically in Fig. 2B. The open channel can either be blocked by quinidine or inactivate. The blocked channel inactivates, rapidly enters the blocked-inactivated state, and subsequently proceeds to the C-type inactivated state. This can be expressed by the following state diagram:
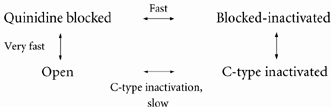 |
(5) |
Inactivation is promoted because the forward rate constants for the Open to Quinidine blocked transition and the Quinidine blocked to Blocked-inactivated transition are much faster than the Open to C-type inactivated transition. Such a scheme also predicts that inactivation decay will be bi-exponential due to there being two pathways out of the open state. Recovery is governed by the direct C-type inactivated to Open transition since dissociation of the drug is probably much faster than recovery from C-type inactivation. However, we cannot measure drug dissociation from C-type inactivated channels, since inactivated channels do not conduct. Recovery from inactivation is the determinant of channel availability, even though the drug may have completely dissociated prior to recovery. Therefore, promotion of C-type inactivation produces ‘use dependence’.
The rate of development of drug-free C-type inactivation is insensitive to voltage at positive potentials. Rapid open channel block of quinidine has a weak voltage dependence corresponding to an effective electrical distance of ≈0.15, which is a result of the need for quinidine to enter the pore and traverse a portion of the electrical field to reach its binding site. We measured the voltage dependence of the rate of development of time-dependent quinidine-promoted C-type inactivation at potentials from +10 to +50 mV (a region over which activation is essentially complete). Data obtained with 800 μm quinidine were fitted with a bi-exponential equation (Fig. 9), and the time constants were plotted against voltage, assuming the following relationship for voltage dependence:
| (6) |
where z, F, R, T and V have their usual meanings, A is gain, and τ is the time constant. The rate constants showed a relatively strong voltage dependence. The effective electrical distance, assuming a monovalent reaction, was 2. This anomalous voltage dependence cannot be accounted for by conventional Woodhull (Woodhull, 1973) analysis and implies a more complex set of physical interactions governing the development of a quinidine-facilitated C-type inactivated state from the simple block of the open channel by quinidine.
Figure 9. The rate of quinidine binding to Kv1.4ΔN is voltage dependent.
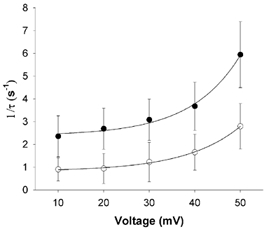
The inactivation rate of the quinidine currents shown in Fig. 8 was best fitted by a bi-exponential function of the form: f =A1exp(-t/τ1) +A2exp(-t/τ2) +C. The two time constants obtained at 800 μm are plotted against voltage (τ1, filled circles; τ2, open circles). The data were fitted to the equation: 1/τ=Aexp((zFδV)/RT)+C, where A is amplitude, V is membrane voltage, δ is the effective electrical distance, z the valency of quinidine (assumed to be 1), R, T and F have their usual meanings, and C is a constant. Data are presented as means ± s.e.m. (n = 3).
Discussion
Ion channels undergo substantial physical rearrangements during gating when they transition between conducting and non-conducting states. These rearrangements can have a strong influence on channel affinity and drug binding, as access to binding sites, and the environment in which they are in, can be altered considerably. Conformation-specific drug-binding properties give rise to clinically important properties, such as use dependence. Models used to describe this behaviour, such as the guarded receptor and modulated receptor models, have viewed gating as something that can affect drug binding (i.e. by revealing a binding site) but cannot itself be affected as a result of drug binding ((Hille, 2001; for review see e.g. Starmer et al. 1984). However, channel gating requires that the pore can open and close rapidly in response to stimuli, and this requires that the two conformations (open and closed) result from a fine energetic balance of hydrophobic and hydrophilic interactions. Drug (or N-terminal) binding in the pore is also a combination of hydrophobic and electrostatic interactions. The mere fact of drug binding changes the hydration energy of the surface of the pore-lining domains which otherwise would be exposed to an aqueous environment. Drug binding can therefore exert a force on the channel. Our finding of an anomalously strong voltage- and time-dependent facet to quinidine block is direct evidence that drug binding is changing the conformational dynamics of the channel. The demonstration of the relationship between drug binding and C-type inactivation is important both for elucidating the mechanism of drug action and for furthering our understanding of the conformational changes that occur in C-type inactivation. Our findings that quinidine affects C-type inactivation in the Kv1.4 channel may apply generally across many voltage-gated ion channels (Rasmusson et al. 1998).
Previous studies have shown that quinidine binds to the open conformation of native and cloned Kv1 channels (Snyders et al. 1992; Yeola et al. 1996), but this study is the first to examine the relationship between quinidine binding and C-type inactivation in a voltage-gated K+ channel. We have previously examined the interaction between C-type inactivation and high-affinity methanesulfonanilide binding to the HERG channel (Wang et al. 1997).
The intracellular binding sites for quinidine have been identified as residues 505 and 512 in the human Kv1.5 channel (Yeola et al. 1996). Assuming that quinidine binds to corresponding residues in Kv1.4 channels, which is likely given the extensive sequence homology in the pore region of the Kv1.4 and Kv1.5 channels, the quinidine binding sites should be located on the intracellular side of the selectivity filter of the channel. Our experiments using the cut-open oocyte technique and studying deactivation tail currents confirm that quinidine binds intracellularly. Quinidine binding to intracellular domains and N-terminal binding during N-type inactivation have the common property that both promote the development of C-type inactivation.
C-type inactivation and drug binding depend heavily on the orientation of side chains on the pore-lining S6 domain (Yeola et al. 1996; Wang et al. 1997; Ficker et al. 1998; Chen et al. 2002; Li et al. 2003). In Kv1.4, the orientation of the open channel that favours drug binding also favours the development of C-type inactivation. Mutations and potassium binding on the extracellular side of the channel alter the open state to prevent both C-type inactivation and rapid open channel block on the intracellular side. This transduction of external actions to an intracellular binding site has been postulated to occur through membrane-spanning domains, particularly S6 (Li et al. 2003).
C-type inactivation can be slowed by extracellular TEA+ or K+ (Hoshi et al. 1990; Bezanilla et al. 1991; Rasmusson et al. 1998; Hille, 2001). Both of these substances affect C-type inactivation in Shaker-type K+ channels by ‘competitive interaction’, i.e. they block the channel and slow inactivation by physically preventing the channel from adopting the C-type inactivated conformation, in a ‘foot-in-the-door’ mechanism (Choi et al. 1991; Demo & Yellen, 1991; Molina et al. 1997).
The dependence of the C-type inactivation rate on [K+]o led to the proposal of two mechanisms to explain how N-terminal domain binding (N-type inactivation) or the binding of an intracellular drug could facilitate C-type inactivation (Rasmusson et al. 1998). The first hypothesis is the Permeation Mechanism (Baukrowitz & Yellen, 1995, 1996a,b), which is based on the idea that blocking the channel prevents K+ permeating through the channel, thus decreasing [K+] in the extracellular vestibule. The reduction in [K+]o means fewer K+ ions bind to the extracellular binding site, so there is no hindrance to the channel adopting the C-type inactivated state (Baukrowitz & Yellen, 1995).
The second hypothesis is the Allosteric Mechanism (Wang et al. 1997; Ficker et al. 1998), which proposes a direct interaction between binding events at the intracellular face and C-type inactivation. Even though C-type inactivation is thought to involve a conformational change at the extracellular mouth of the pore, the channel must be regarded as an integrated unit in conformational terms, i.e. a conformational change at a remote site may result in allosteric changes throughout the protein. This leads to the result that an action at the intracellular face of the channel such as quinidine or N-terminal binding can affect an extracellular event such as C-type inactivation. Transmembrane communication with allosteric mechanisms connecting physically remote sites has already been demonstrated for Kv1.4 channels, e.g. mutating a valine at the C-terminal end of S6 has a dramatic effect on C-type inactivation of the Kv1.4 channel (Li et al. 2003). This kind of transmembrane communication is difficult to reconcile with the permeation mechanism.
Our study demonstrates that two modifications to the extracellular face of the channel (i.e. elevation of extracellular [K+] and the K532Y mutation) affect the binding of quinidine at the intracellular side of the pore. Furthermore, the binding of quinidine to the intracellular side enhances C-type inactivation, which is an extracellular phenomenon.
Elevation of [K+]o can reduce the affinity for drug binding by a direct (electrostatic or knock-off) effect or by an indirect effect via modification of the pore region by some kind of conformational change (allosteric effect), or by a combination of both (Wang et al. 1997). If the direct electrostatic effect was the basis of the interaction, elevating [K+]o should reduce quinidine binding to a similar degree in both fKv1.4ΔN and fKv1.4[K532Y]ΔN channels. Our data show that increasing [K+]o had very different results on these two channels (elevated [K+]o reduced the affinity of quinidine for fKv1.4ΔN, but had no effect in fKv1.4[K532Y]ΔN channels), which indicates that the modulation of drug binding by [K+]o is not the result of an electrostatic interaction. The only other influence [K+]o has on the channel is the conformational change following binding of K+ to the extracellular binding site.
Several other results in this paper are difficult to reconcile with the permeation mechanism.
(1) [K+]o accumulation near the extracellular mouth will be largest at positive potentials, where there is most current flow. The permeation mechanism would therefore predict that C-type inactivation is slowest at positive potentials. However, C-type inactivation is voltage insensitive at positive potentials and slower near threshold (Rasmusson et al. 1995a).
(2) The permeation mechanism predicts that rapid quinidine open channel block will be independent of [K+]o accumulation because [K+]o is presumed to affect only the development of C-type inactivation, and not the quinidine binding site. The connection between the rapid quinidine binding affinity and [K+]o is consistent with an allosteric linkage between the binding of quinidine and the development of C-type inactivation in which the two processes are energy additive.
(3) The strong voltage dependence (and effective valency of 2) for the development of the quinidine-induced C-type inactivated state is too large to be accounted for by conventional models involving the permeation pathway. The KcsA crystal structure suggests that the permeation pathway has at most partial occupancy by two potassium ions, so the effective valency must be considerably less than 2 (Doyle et al. 1998). The amount of the field experienced by either potassium ion in the selectivity filter must be considerably less than 100 % of the total transmembrane field. Furthermore, the exit of the first ion must occur on a time scale of microseconds for permeation to occur at all. This leaves the maximum voltage dependence for the ‘last ion to exit the pore’ to be less than 1 (Baukrowitz & Yellen, 1996b) in the permeation model. In other words, the voltage dependence of quinidine-induced C-type inactivation is too strong to be accounted for by charge within the permeation pathway and probably involves charged residues on the transmembrane domains.
These data show there is a link between C-type inactivation-mediated pore closure and the quinidine-binding sites in the intracellular vestibule region. Since the orientation of S6 is critical to drug binding (Chen et al. 2002), and recent studies have shown a link between this domain and C-type inactivation (Li et al. 2003), this suggests that S6 moves during C-type inactivation. Therefore, the conformational change accompanying C-type inactivation includes more of the channel than previously thought and has an allosteric effect on the intracellular quinidine-binding site.
We interpreted our data in the context of the allosteric model. Quinidine binds rapidly to the open channel. We propose that the orientation of S6 that forms the high affinity quinidine-binding site in the open channel is closely related to the orientation of S6 that leads to the C-type inactivated state (Chen et al. 2002; Li et al. 2003). Binding of quinidine to the open state results in a rearrangement of the open channel into the C-type inactivated state, producing the time-dependent component of block. C-type inactivation involves a conformational change, and manipulations that prevent the development of C-type inactivation must therefore affect the likelihood of the channel adopting certain conformations. This is consistent with the idea that interfering with C-type inactivation makes the quinidine-bound state less energetically favourable. Therefore, manipulations that reduce C-type inactivation also reduce open channel quinidine block. When the channel is blocked by quinidine, which is bulky, lipophilic, and positively charged, physical interactions will deform the channel. We propose that deformation following the initial open channel block gives rise to the voltage dependence of the slow time-dependent component of block.
Mathematical modelling of quinidine block
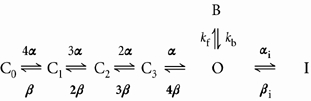
In order to test our hypothesis that quinidine binding drives the channel into a drug-bound C-type inactivated state, we constructed a mathematical model of quinidine binding to fKv1.4ΔN. A simple model of a channel undergoing open channel block can be represented by the following state model:
 |
(7) |
where C0 to C3 represent voltage-dependent closed states which must be traversed during opening of the channel in response to a depolarising change in potential, O is the open conducting state, B is a drug-blocked state, and α and β are rate constants defined in Methods. After activation is complete, drug binding is at its steady-state level, and the number of channels in the open and blocked states is in equilibrium. The rate constants for the activation gating process were derived from Comer et al. (1994). The rate constants for open channel block, kf and kb, have a weak voltage dependence corresponding to the measured effective electrical distance and are given by the equations:
| (8) |
| (9) |
where [D] is the quinidine concentration, KD is the quinidine dissociation constant at 2 mm [K+]o, and V is voltage.
The rate constants for rapid open channel block are beyond the resolution of our kinetic measurements and are not separable from activation. We assumed a scaling factor that was equal to the activation rate at +50 mV for the forward rate constant, kf. The backward rate constant, kb was calculated from the equilibrium block at +50 mV. This simple model (Fig. 10) reproduces the peak I-V relationship and cross-over tail currents shown in Fig. 1C. It also reproduces the effective electrical distance measurements.
Figure 10. A model of rapid open channel block by quinidine.
Simulated fKv1.4ΔN currents were calculated for a series of depolarising pulses from −90 to +50 mV in 10 mV increments with 2 mm[K+]o. Currents were recalculated to simulate the block by 100 μm quinidine. A, peak current-voltage relationships in control and 100 μm quinidine. Currents were normalised to the maximum peak current obtained at +50 mV under control conditions, to enable comparison with experimental data in Fig. 1. B, simulated tail currents exhibit the ‘cross-over’ seen experimentally. Currents were calculated in response to a repolarising pulse from +50 to −120 mV to generate deactivation tail currents in the presence or absence of 80 μm quinidine. The characteristic slowing of deactivation shown experimentally in Fig. 1 is reproduced. C, simple open channel block predicts a slowing of C-type inactivation in the presence of quinidine.
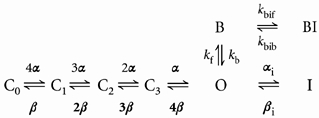
The simple model of open channel block given in eqn (7) was modified to incorporate voltage-insensitive C-type inactivation:
 |
(10) |
where αi and βi are the inactivation rates. A major shortcoming of this is that inactivation and drug binding compete. Even if drug binding (kf) occurs on a time scale that is orders of magnitude faster than C-type inactivation (αi), in the simple model of eqn (10), the rate of development of C-type inactivation is always slowed in the presence of the drug (Fig. 10C). In this type of model, drug binding lowers the probability of the channel being in O, the open state. This model cannot explain our experimental results, which clearly show that quinidine increases the C-type inactivation rate.
Following rapid open channel block, the channel changes in a time-dependent manner to a blocked-inactivated state:
 |
(11) |
where kbif is the forward rate constant for the transition from B to BI and kbib is the backward rate constant for the transition from BI to B. With this model, inactivation during a single depolarising pulse will exhibit both a rapid open channel block and development of a slower inactivated state. If the transition from B, the blocked state, to BI, the blocked-inactivated state, includes a voltage-sensitive on-rate then the voltage-dependent increase in current decay is also reproduced. However, this model still results in a bi-exponential decay with one component slowed relative to C-type inactivation in the absence of quinidine and with a bi-exponential recovery that reflects the differing properties of the drug-bound inactivated state and the C-type inactivated state. In order to have a model that produces an increase in the rate of inactivation in the presence of drug and a single pathway for recovery, we added a final set of transitions to link the blocked-inactivated state and the C-type inactivated state:
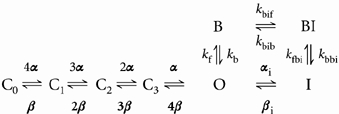 |
(12) |
where kfbi is the rate constant for the transition from I to BI and kbbi is the rate constant for the transition from BI to I. In this model, the inactivation rate is increased in the presence of quinidine and is voltage dependent (Fig. 11). Recovery from inactivation is dependent mainly on the ‘normal’ rate of recovery from C-type inactivation (Fig. 12). This normal recovery explains a potential paradox in our data. Quinidine clearly has strong interactions with the C-type inactivated state at positive potentials. Regardless of the model of gating, this free energy should contribute to the stability of the drug-bound C-type inactivated state and transitions into and out of that state.
Figure 11. A model of rapid open channel block by quinidine which develops into C-type inactivation.
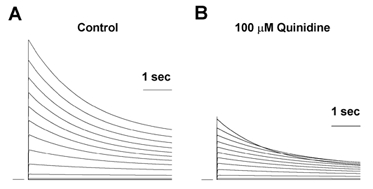
A, simulated fKv1.4ΔN currents were calculated for a series of depolarising pulses from −90 to +50 mV in 10 mV increments with 2 mm[K+]o. B, currents were recalculated to simulate the block by 100 μm quinidine. Results are shown as peak current-voltage relationships. Currents were normalised to the maximum peak current obtained at +50 mV under control conditions, to enable comparison with experimental data in Fig. 1.
Figure 12. Simulation of the effect of quinidine on the rate of recovery from inactivation.
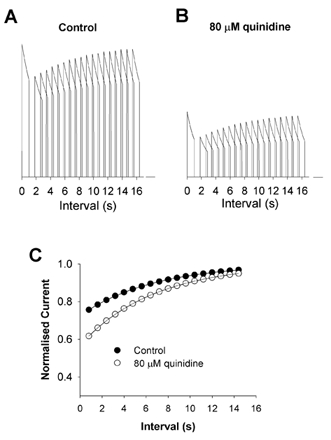
Protocols were the same as described in Fig. 8. A, control simulations. B, simulations in the presence of 80 μm quinidine. C, peak currents during P2 normalised to peak P1 currents and plotted as a function of Δt. The filled and open circles represent control and quinidine data, respectively. Data were fitted using the equation: f = 1 – (Aexp(-t/τ)), where f is fractional current, A is the amplitude of the current, t is duration and τ is the time constant.
It may seem paradoxical that quinidine has a strong effect on the development of C-type inactivation while the kinetics of recovery are relatively unaffected. However, this can be explained by considering the voltage dependence of the rate constants of inactivation and recovery. The channel enters the inactivated state when the membrane is depolarised. When quinidine is present, the free energy of the interaction between quinidine binding and the C-type inactivated state will contribute to the stability of the drug-bound C-type inactivated state and the transitions into and out of that state (which is independent of the model of gating used to describe the process). Our data show that entry into the quinidine-induced inactivated state is strongly voltage sensitive. At positive potentials, quinidine binding has strong interactions with the C-type inactivated state, promoting its development. This explains the strong effect of quinidine on the development of C-type inactivation at positive potentials. Recovery is a process that occurs at hyperpolarised potentials. Transitions from the blocked to blocked-inactivated channel have an effective valency of 2, so it is energetically unfavourable for the channel to exit the inactivated state via the blocked-inactivated- blocked- open pathway at hyperpolarised potentials (see eqn (12)). Therefore, the channel in the presence of quinidine recovers through the same pathway (inactivated- open-closed) as the control channel does.
Conclusion
Quinidine block of Kv1.4ΔN channels is a complex process. Rapid open channel block is just the first step in forming a long-lived quinidine-induced inactivated state. This quinidine-induced inactivation process has an anomalous voltage dependence which does not arise from a conventional Woodhull (Woodhull, 1973) type of model, but is likely to be the result of quinidine-induced changes in protein conformation. These changes in conformation and their voltage dependence are important determinants of the recovery of channels from the quinidine-induced inactivated state.
Acknowledgments
This work was supported in part by grants from the NIH NHLBI, HL-59526 (R.L.R.), HL-52874 (H.C.S.), HL-19216 (H.C.S.), NSF KDI grant DBI-9873173 (R.L.R.) and an Established Investigator Award from the American Heart Association (R.L.R.).
References
- Balser JR, Bennett PB, Hondeghem LM, Roden DM. Suppression of time-dependent outward current in guinea pig ventricular myocytes. Actions of quinidine and amiodarone. Circ Res. 1991;69:519–529. doi: 10.1161/01.res.69.2.519. [DOI] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Chiamvimonvat N, Pérez-García MT, Marban E, Tomaselli GF. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol. 1996;494:431–442. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Two functionally distinct subsites for the binding of internal blockers to the pore of voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1996a;93:13357–13361. doi: 10.1073/pnas.93.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996b;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Rasmusson RL. Models of cardiac ion channels. In: Cabo C, Rosenbaum DS, editors. Quantitative Cardiac Electrophysiology. NY, USA: Marcel Dekker, Inc.; 2002. pp. 1–60. [Google Scholar]
- Bezanilla F, Perozo E, Papazian DM, Stefani E. Molecular basis of gating charge immobilization in Shaker potassium channels. Science. 1991;254:679–683. doi: 10.1126/science.1948047. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Trimmer JS, Nerbonne JM, Strauss HC. Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel alpha subunits in ferret left ventricular myocytes. J Gen Physiol. 1999;113:581–600. doi: 10.1085/jgp.113.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FS, Fedida D. On the mechanism by which 4-aminopyridine occludes quinidine block of the cardiac K+ channel, hKv1. 5. J Gen Physiol. 1998;111:539–554. doi: 10.1085/jgp.111.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Sanchez-Chapula J, Salinas-Stefanon E, Duff HJ, Giles WR. Quinidine-induced open channel block of K+ current in rat ventricle. Br J Pharmacol. 1995;115:335–343. doi: 10.1111/j.1476-5381.1995.tb15882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer MB, Campbell DL, Rasmusson RL, Lamson DR, Morales MJ, Zhang Y, Strauss HC. Cloning and characterization of an Ito-like potassium channel from ferret ventricle. Am J Physiol. 1994;267:H1383–1395. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais CJ, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fedida D. Gating charge and ionic currents associated with quinidine block of human Kv1. 5 delayed rectifier channels. J Physiol. 1997;499:661–675. doi: 10.1113/jphysiol.1997.sp021959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of HERG K+ channels. Circ Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates Inc.; 2001. [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Smith PL, Yellen G. Trapping of organic blockers by closing of voltage-dependent K+ channels: evidence for a trap door mechanism of activation gating. J Gen Physiol. 1997;109:527–535. doi: 10.1085/jgp.109.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y, Giles WR. Quinidine-induced inhibition of transient outward current in cardiac muscle. Am J Physiol. 1987;253:H704–708. doi: 10.1152/ajpheart.1987.253.3.H704. [DOI] [PubMed] [Google Scholar]
- Li XY, Bett GCL, Jiang XJ, Bondarenko VE, Morales MJ, Rasmusson RL. Regulation of N- and C-type inactivation by pHo and potassium in normal and mutant Kv1. 4 channels: Evidence for transmembrane communication. Am J Physiol Heart Circ Physiol. 2003;284:H71–80. doi: 10.1152/ajpheart.00392.2002. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- McKinnon D. Molecular identity of Ito: Kv1. 4 redux. Circ Res. 1999;84:620–622. doi: 10.1161/01.res.84.5.620. [DOI] [PubMed] [Google Scholar]
- Molina A, Castellano AG, Lopez-Barneo J. Pore mutations in Shaker K+ channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J Physiol. 1997;499:361–367. doi: 10.1113/jphysiol.1997.sp021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Castellino RC, Zhang Y, Campbell DL, Strauss HC. C-type inactivation controls recovery in a fast inactivating cardiac K+ channel (Kv1. 4). expressed in Xenopus oocytes. J Physiol. 1995a;489:709–721. doi: 10.1113/jphysiol.1995.sp021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- Rasmusson RL, Zhang Y, Campbell DL, Comer MB, Castellino RC, Liu S, Strauss HC. Bi-stable block by 4-aminopyridine of a transient K+ channel (Kv1. 4) cloned from ferret ventricle and expressed in Xenopus oocytes. J Physiol. 1995b;485:59–71. doi: 10.1113/jphysiol.1995.sp020712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawsky MT, Castle NA. K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. J Pharmacol Exp Ther. 1994;269:66–74. [PubMed] [Google Scholar]
- Snyders DJ, Knoth KM, Roberds SL, Tamkun MM. Time-, voltage-, and state-dependent block by quinidine of a cloned human cardiac potassium channel. Mol Pharmacol. 1992;41:322–330. [PubMed] [Google Scholar]
- Snyders DJ, Yeola SW. Determinants of antiarrhythmic drug action. Electrostatic and hydrophobic components of block of the human cardiac hKv1.5 channel. Circ Res. 1995;77:575–583. doi: 10.1161/01.res.77.3.575. [DOI] [PubMed] [Google Scholar]
- Starmer CF, Grant AO, Strauss HC. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys J. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todt H, Dudley SC, Jr, Kyle JW, French RJ, Fozzard HA. Ultra-slow inactivation in mu1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys J. 1999;76:1335–1345. doi: 10.1016/S0006-3495(99)77296-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GN. Molecular structure of cardiac Ito channels: Kv4. 2, Kv4.3, and other possibilities? Cardiovasc Res. 1999;41:16–18. doi: 10.1016/s0008-6363(98)00282-x. [DOI] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett. 1997;417:43–47. doi: 10.1016/s0014-5793(97)01245-3. [DOI] [PubMed] [Google Scholar]
- Wang SY, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ Res. 1999;84:551–561. doi: 10.1161/01.res.84.5.551. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Jegla TJ, Kaprielian R, Backx PH. Regional contributions of Kv1. 4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. Am J Physiol. 1999;276:H1599–1607. doi: 10.1152/ajpheart.1999.276.5.H1599. [DOI] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- Yang T, Snyders DJ, Roden DM. Inhibition of cardiac potassium currents by the vesnarinone analog OPC-18790: comparison with quinidine and dofetilide. J Pharmacol Exp Ther. 1997;280:1170–1175. [PubMed] [Google Scholar]
- Yatani A, Wakamori M, Mikala G, Bahinski A. Block of transient outward-type cloned cardiac K+ channel currents by quinidine. Circ Res. 1993;73:351–359. doi: 10.1161/01.res.73.2.351. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Snyders DJ. Electrophysiological and pharmacological correspondence between Kv4. 2 current and rat cardiac transient outward current. Cardiovasc Res. 1997;33:540–547. doi: 10.1016/s0008-6363(96)00221-0. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhu B, Yao JA, Tseng GN. Differential effects of S6 mutations on binding of quinidine and 4-aminopyridine to rat isoform of Kv1. 4: common site but different factors in determining blockers' binding affinity. J Pharmacol Exp Ther. 1998;287:332–343. [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]