

Abstract
Hypoxia-evoked vasodilatation is a fundamental regulatory mechanism that is often attributed to adenosine. The identity of the O2 sensor is unknown. Nitric oxide (NO) inhibits endothelial mitochondrial respiration and ATP generation by competing with O2 for its binding site on cytochrome oxidase. We proposed that in vivo this interaction allows endothelial cells to release adenosine when O2 tension falls or NO concentration increases. Using anaesthetised rats, we confirmed that the increase in femoral vascular conductance (FVC, hindlimb vasodilatation) evoked by systemic hypoxia is attenuated by NO synthesis blockade with l-NAME, but restored when baseline FVC is restored by infusion of NO donor. This ‘restored’ hypoxic response, like the control hypoxic response, is inhibited by the adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine. Similarly, the FVC increase evoked by adenosine infusion was attenuated by l-NAME but restored by infusion of NO donor. However, when baseline FVC was restored after l-NAME with 8-bromo-cGMP, the FVC increase evoked by adenosine infusion was restored, but not in response to systemic hypoxia, suggesting that adenosine was no longer released by hypoxia. Infusion of NO donor at a given rate after treatment with l-NAME evoked a greater FVC increase during systemic hypoxia than during normoxia, both responses being reduced by 8-cyclopentyl-1,3-dipropylxanthine. Finally, both bradykinin and NO donor released adenosine from superfused endothelial cells in vitro; l-NAME attenuated only the former response. We propose that in vivo, shear-released NO increases the apparent Km of endothelial cytochrome oxidase for O2, allowing the endothelium to act as an O2 sensor, releasing adenosine in response to moderate falls in O2.
Systemic vascular regions respond to a decrease in blood O2 concentration with a compensatory vasodilatation that restores the O2 supply. The mechanism by which this decrease in O2 tension (PO2) is sensed has not yet been established. However, it is thought that compromised ATP synthesis results in the release of adenosine, which is intimately involved in the hypoxia-evoked vasodilatation of several different vascular beds (Berne, 1980; Neylon & Marshall, 1991; Nakhostine & Lamontagne, 1993; Armstead, 1997; Edmunds & Marshall, 2001b).
Adenosine might be released from a range of cell types including the tissue parenchymal cells, the sympathetic nerve fibres and the vascular endothelium. During systemic hypoxia, the available evidence indicates that in resting skeletal muscle, the majority of the adenosine is released from vascular endothelium and that it acts on endothelial A1 receptors to induce vasodilatation (see Marshall, 2000). Indeed, in a recent study, involving the microdialysis technique, systemic hypoxia had no detectable effect on the adenosine concentration measured in the interstitial fluid of skeletal muscle, but increased that measured in the venous efflux, consistent with endothelial or vascular release of adenosine. By contrast, during muscle contraction, adenosine concentration increased in both the interstitial fluid and the venous efflux, indicating release predominantly from the skeletal muscle fibres (Mo & Ballard, 2001). Even in a beating heart preparation, hypoxic perfusion of the coronary circulation, which would have been expected to compromise oxidative metabolism of the cardiac myocytes, caused an 11-fold increase in the release of radiolabelled adenosine that had been preferentially loaded into the endothelium, as well as a ninefold increase in the release of unlabelled adenosine, mainly from the cardiac myocytes (Deussen et al. 1986). It has been proposed that during systemic hypoxia in resting skeletal muscle, the vasodilatation and increased O2 delivery caused by adenosine release from the endothelium protects the skeletal muscle fibres against hypoxia unless the reduction in O2 in the blood is very severe (Marshall, 2000).
Tissue PO2 within resting skeletal muscle averages 15-20 mmHg, and rarely falls below 5 mmHg, even in local regions of muscle, during severe systemic hypoxia (Harrison et al. 1990). However, the critical tissue PO2 at which mitochondrial oxidative metabolism is limited appears to be < 1 mmHg in vitro (Wilson et al. 1973; Rosenthal et al. 1976; Jöbsis, 1977) and as low as ≈5 mmHg muscle in vivo (Conley et al. 2000). Therefore these findings are not consistent with the metabolic release of adenosine from either the vasculature or muscle during systemic hypoxia. However, nitric oxide (NO) can compete with O2 at the O2-binding site of cytochrome oxidase (complex IV of the mitochondrial respiratory chain), and by doing so it increases the apparent Km of the enzyme for O2 (Brown & Cooper, 1994; Cleeter et al. 1994; Schweizer & Richter, 1994). In perfused endothelial cells in vitro, endogenously released NO has been shown to downregulate O2 consumption, especially at low O2 concentrations, leading to the suggestion that an interaction between NO and O2 at the level of cytochrome oxidase allows it to act as an O2 sensor (Clementi et al. 1999). Thus, it is a reasonable hypothesis that in the presence of endogenous, shear-released NO in vivo, inhibition of endothelial mitochondrial respiration can occur following a relatively small decrease in PO2. If so, this would allow endothelial cells to release adenosine and so initiate vasodilatation.
To test this hypothesis we have used in vitro and in vivo methods to investigate the dependence on NO of endothelial release of adenosine during normoxia and hypoxia.
Methods
In vivo experiments
In male Wistar rats (200-250 g), anaesthesia was induced with an O2-halothane mixture (3.5 % halothane) and maintained with Saffan (Schering-Plough Animal Health, Welwyn Garden City, UK) delivered at 5-12 mg kg−1 h−1, i.v. Arterial blood pressure (ABP), femoral blood flow and femoral vascular conductance (FVC) were recorded as described previously (Bryan & Marshall, 1999a; Edmunds & Marshall, 2001a, b). The animals breathed spontaneously throughout the experiment. All experiments were performed in accordance with the UK Animals (Scientific Procedures) Act 1986. At the end of the experiments, the animals were killed with an overdose of anaesthetic.
Protocols
In group 1 (n = 9), after a 25 min equilibration period, the inspirate was changed from air to 12 % or 8 % O2 for 5 min periods each by changing the N2/O2 mixture blown across the side arm of the tracheal cannula (see Edmunds & Marshall, 2001a). A 15 min recovery period was allowed between the hypoxic challenges. The NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME; 10 mg kg−1, i.v.; see Bryan & Marshall, 1999b) was then given, which decreased baseline FVC (Fig. 1), and approximately 30 min later the hypoxic challenges were repeated. Baseline FVC was then restored by infusion of S-nitroso-N-acetylpenicillamine (SNAP; 10 μg kg−1 min−1, i.a.), and when variables were stable the hypoxic challenges were repeated as above. Finally, the adenosine A1 receptor antagonist 8-cyclopentyl-1, 3-dipropylxanthine (DPCPX; 0.1 mg kg−1, i.v., Bryan & Marshall, 1999a) was given while the SNAP infusion continued, and 20 min later the hypoxic challenges were repeated.
Figure 1. After inhibition of nitric oxide synthase (NOS), S-nitroso-N-acetylpenicillamine (SNAP) infusion restores the hypoxia-evoked vasodilatation mediated by adenosine.

Each column shows femoral vascular conductance (FVC ± s.e.m.) during air breathing (□) and at the 5th min of breathing 12 % O2 ( ). From left to right: under control conditions, after l-NAME, after l-NAME during SNAP infusion, after l-NAME and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) during continuing SNAP infusion. Symbols below columns (###) indicate differences between baseline FVC values during air breathing, symbols above columns (***) indicate differences between changes in FVC recorded in hypoxia; P < 0.001 for both * and #. Arterial blood pressure (ABP) and femoral blood flow (FBF) values recorded during these experiments are available as Table 1 in the online version of this paper at:
). From left to right: under control conditions, after l-NAME, after l-NAME during SNAP infusion, after l-NAME and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) during continuing SNAP infusion. Symbols below columns (###) indicate differences between baseline FVC values during air breathing, symbols above columns (***) indicate differences between changes in FVC recorded in hypoxia; P < 0.001 for both * and #. Arterial blood pressure (ABP) and femoral blood flow (FBF) values recorded during these experiments are available as Table 1 in the online version of this paper at:
In group 2 (n = 10), essentially the same protocol was followed except that in addition to the hypoxic stimuli, a 5 min adenosine infusion (1.2 mg kg−1 min−1, i.a.) was given after l-NAME, and baseline FVC was restored with 8-bromo-cGMP (25 mg kg−1 h−1, i.a.) rather than SNAP. Hypoxic challenges and adenosine infusions were repeated.
Group 3 (n = 10) was given l-NAME (10 mg kg−1) to inhibit any endogenous production of NO. SNAP (20 μg kg−1 min−1) was infused for a 5 min period during air breathing and when FVC had stabilised during a period of hypoxia (12 % O2). Following another recovery period, DPCPX (0.1 mg kg−1, i.v) was administered and the protocol was repeated.
Endothelial cell culture and utilisation
Endothelial cells, prepared from fresh porcine thoracic aortae obtained from the abattoir, were cultured initially in Dulbecco's modified Eagle's medium plus 20 % fetal calf serum, and were then grown to confluence on microcarrier beads under mild periodic stirring (Gryglewski et al. 1986; Clementi et al. 1999). After 7 days, preparations in which < 90 % of the beads were covered by confluent cells were discarded.
Endothelial-cell-coated beads were packed into a column and perfused, at a rate of 2 ml min−1, with Krebs-Henseleit solution of the following composition (mm): NaCl 136.9, KCl 4.7, CaCl2 1.8, MgSO4 1.1, KH2PO4 1.2, NaHCO3 25, glucose 5 and pyruvate 2 (37 °C, pH 7.4, equilibrated at PO2 110 mmHg). Also included in the Krebs-Henseleit solution was dipyridamole, 1 μm, to inhibit adenosine uptake into endothelial cells, and erythro-9-(2-hydroxy-3-nonyl)-adenine, 5 μm, to inhibit adenosine deaminase. Cell viability was confirmed by the measurement of bradykinin-induced release of NO, determined by bioassay (Gryglewski et al. 1986). The NO donor diethyltriamine NONOate (DETA-NO), and bradykinin were then given as bolus injections over a 30 s period to give final concentrations of 0.5 mm and 1 μm, respectively. Perfusate samples were then taken for analysis of adenosine concentration by HPLC (Zhang et al. 1991). An injection volume of 100 μl and a flow rate of 1 ml min−1 were used. The retention time was 7.5 min.
Statistical analysis of data
All data are expressed as means ± s.e.m. Changes in FVC seen during hypoxia were computed as changes in the FVC integral and are expressed in conductance units (CU). Differences were determined with repeated-measures ANOVA. When global ANOVA showed P < 0.05, Fisher's post hoc test was performed where P < 0.05 was considered significant.
Results
In group 1, systemic hypoxia (12 % inspired O2: arterial PO2 ≈40 mmHg; Neylon & Marshall, 1991) evoked a fall in ABP, no significant change in femoral blood flow and an increase in FVC, demonstrating vasodilatation in the hindlimb vasculature, as described previously (Neylon & Marshall, 1991; Bryan & Marshall, 1999a; Edmunds & Marshall, 2001a, b). l-NAME not only increased baseline ABP and reduced baseline FVC, but also severely attenuated the hypoxia-evoked vasodilatation (Fig. 1; see Bryan & Marshall, 1999b). When ABP and baseline FVC were restored with SNAP, the vasodilator response to hypoxia was also completely restored (Fig. 1). This vasodilatation was reduced by ≈50 % by the A1 antagonist DPCPX (see Edmunds & Marshall, 2001b). Similar results were obtained at the more severe level of hypoxia (arterial PO2 ≈34 mmHg; Neylon & Marshall, 1991) produced by inspiring 8 % O2 (data not shown). These data confirm previous results obtained using sodium nitroprusside (SNP) as a NO donor (Edmunds & Marshall, 2001a). SNP could not be used for the purposes of the present study due to its cyanide-mediated effects on mitochondrial respiration (Norris & Hume, 1987).
In group 2, adenosine was infused at a rate selected to cause an increase in FVC that mimicked the contribution of adenosine to the hypoxia-evoked vasodilatation under normal conditions (≈50 % of the total increase in FVC; Bryan & Marshall, 1999a). Like systemic hypoxia, adenosine infusion also evoked a fall in ABP (see Bryan & Marshall, 1999a). As expected (see Bryan & Marshall, 1999b), l-NAME attenuated the responses to both hypoxia and adenosine (Fig. 2). However, when baseline FVC was then restored by infusion of the membrane-permeable analogue of cGMP, 8-bromo-cGMP, the vasodilatation evoked by hypoxia remained severely blunted, whereas the vasodilatation evoked by exogenous adenosine was restored (Fig. 2).
Figure 2. Infusion of 8-bromo-cGMP (8-Br-cGMP) restores adenosine- but not hypoxia-evoked hindlimb vasodilatation.
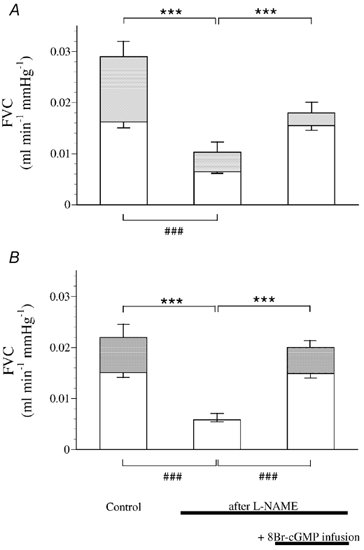
Each column shows FVC (± s.e.m.) during air breathing (□), and at the 5th minute of breathing 12 % O2 (A; ) or following adenosine infusion (B;  ). From left to right in A and B: under control conditions, after l-NAME and after l-NAME during infusion of 8-Br-cGMP. Symbols below columns (###) indicate differences between baseline FVC values during air breathing, symbols above columns (***) indicate differences between evoked changes in FVC; P < 0.001. ABP and FBF values recorded during these experiments are available as Table 2 in the online version of this paper at:
). From left to right in A and B: under control conditions, after l-NAME and after l-NAME during infusion of 8-Br-cGMP. Symbols below columns (###) indicate differences between baseline FVC values during air breathing, symbols above columns (***) indicate differences between evoked changes in FVC; P < 0.001. ABP and FBF values recorded during these experiments are available as Table 2 in the online version of this paper at:
In group 3, infusion of SNAP at a given rate (see Methods) caused an increase in FVC that was greater during systemic hypoxia than during normoxia (Fig. 3). Furthermore, DPCPX attenuated the vasodilatation evoked by SNAP under both conditions. Following treatment with DPCPX, there was no significant difference in the vasodilator actions of SNAP during normoxia and hypoxia.
Figure 3. Hindlimb vasodilatation evoked by SNAP in the presence of NOS inhibition is greater during hypoxia than during normoxia and is sensitive to adenosine A1-receptor inhibition under both conditions.

Columns show ‘baseline’ FVC values (± s.e.m.) during air breathing (□), during hypoxia (12 % O2; ) and at the 5th min of SNAP infusion (▪). All tests were made in the presence of l-NAME; DPCPX was given between columns 2 and 3, as indicated below columns. Symbols below columns (###) indicate differences between FVC values recorded during normoxia and hypoxia; P < 0.001. **Significant difference between changes evoked in FVC by SNAP in normoxia and hypoxia; P < 0.01. ††Significant difference between changes evoked in FVC by SNAP in normoxia before and after DPCPX; P < 0.01. §§Significant difference between changes evoked in FVC by SNAP in hypoxia before and after DPCPX; P < 0.01. For ABP and FBF values recorded during these experiments are available as Table 3 in the online version of this paper at:
Both the NO donor DETA-NO and bradykinin caused a significant release of adenosine from endothelial cells in vitro (Fig. 4). l-NAME did not inhibit the adenosine release evoked by DETA-NO, but abolished that evoked by bradykinin (Fig. 4).
Figure 4. Both endogenously produced nitric oxide (NO) and exogenous NO can stimulate adenosine release from cultured endothelial cells.
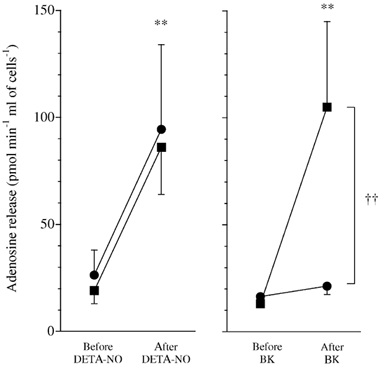
Peak adenosine release from endothelial cells within 5 min of diethyltriamine NONOate (DETA-NO; left, n = 8) or bradykinin (BK; right, n = 8), before (▪) and after (•) treatment with l-NAME (100 μm). **P < 0.01 before vs after DETA-NO or BK. ††P < 0.01 before vs after l-NAME.
Discussion
In this study, we confirm that inhibition of NOS with l-NAME substantially attenuates the hindlimb muscle vasodilatation (increase in FVC) evoked both by adenosine infusion and systemic hypoxia (see Bryan & Marshall, 1999a). Moreover, when FVC was restored after l-NAME by infusion of the NO donor SNAP, the hypoxia-evoked vasodilatation was also restored and this subsequent vasodilatation was sensitive to the A1 receptor antagonist DPCPX. We have already shown that when baselines are restored after treatment with l-NAME by infusion of the prostacyclin analogue iloprost rather than an NO donor, the hypoxia-induced dilatation remains attenuated (Edmunds & Marshall, 2001a). Thus, our data confirm that NO is required for hypoxia-induced hindlimb vasodilatation, and that this vasodilatation can be specifically restored by exogenous NO when NOS is inhibited. They also confirm that adenosine acting on A1 receptors is a main contributor to this ‘restored’ dilatation, as it is to the vasodilatation evoked by systemic hypoxia in the absence of l-NAME (see Bryan & Marshall, 1999b; Edmunds & Marshall, 2001a, b). Therefore, we surmised that either the vasodilator actions of adenosine and/or the release of adenosine in hypoxia are dependent on the presence of endogenous NO.
The classically recognised second messenger for NO is cGMP. It has been demonstrated that cGMP sensitises vascular smooth muscle to the actions of other vasodilators, at least in part by inhibiting smooth muscle phosphodiesterase (PDE): PDE 3 (Delpy et al. 1996). Thus, we investigated whether the dilatation evoked by systemic hypoxia or by infusion of adenosine required the NO-dependent generation of cGMP. In fact, when the baseline level of FVC was restored after l-NAME with 8-bromo-cGMP, the hypoxia-evoked vasodilatation remained severely blunted, but the hindlimb vasodilatation evoked by adenosine infusion was restored. Adenosine is known to cause vasodilatation by stimulating A2A receptors and increasing cAMP levels in vascular smooth muscle (Olsson & Pearson, 1991). There is also evidence to suggest that adenosine can cause vasodilatation by acting on vascular smooth muscle A1 receptors to open ATP-sensitive K+ channels (Dart & Standen, 1993), or by acting on A1 or A2A receptors on the endothelium to increase the synthesis of NO (Sobrevia et al. 1997; Li et al. 1998; Ray et al. 2002). In hindlimb muscle, exogenous adenosine evokes vasodilatation by stimulating both A1 and A2A receptors, both of these actions being largely NO-dependent (Bryan & Marshall, 1999a, b). Thus, when NO synthesis was blocked by l-NAME, it is likely that 8-bromo-cGMP restored the dilator response to exogenous adenosine by restoring its facilitatory interaction with cAMP. This being the case, if adenosine were released during hypoxia in the presence of l-NAME and SNAP, it should have been able to cause vasodilatation, as indeed it could when baseline FVC was restored with SNAP. In other words, we can propose that the vasodilator action of adenosine, whether endogenous or exogenous, requires the presence of NO acting at the soluble guanylate cyclase, or a tonic level of cGMP. By contrast, the release of adenosine during hypoxia is apparently dependent on the presence of NO, but independent of the actions of NO on guanylate cyclase. Given the evidence that the adenosine that is vasoactive in systemic hypoxia is released from the endothelial cells rather than the skeletal muscle fibres (see Marshall, 2000; Mo & Ballard, 2001), it is reasonable to further propose that it is the endothelial release of adenosine that is dependent on the presence of endogenous NO.
Consumption of O2 by endothelial cells that are superfused at physiological levels of PO2, and therefore subjected to shear stress, is modulated by endogenously produced NO such that inhibition of NOS increases their O2 consumption (Clementi et al. 1999). These studies also demonstrate that endothelial cells are metabolically active and respire at significant rates. In view of the competitive nature of the interaction between NO and O2 under physiological conditions, when O2 levels remain constant, any increase in NO concentration should have the same effect on the endothelial cells as a decrease in the concentration of O2. This was why we investigated the involvement of adenosine in the vasodilator actions of SNAP in anaesthetised rats breathing either room air or a hypoxic gas mixture (12 % inspired O2). Our results showed that SNAP causes an increase in FVC that is greater during hypoxia than during normoxia. Furthermore, DPCPX inhibited the vasodilatation evoked by SNAP under both conditions. Thus, these results indicate, for the first time, that adenosine can be released by an NO donor and contribute significantly to NO-induced vasodilatation by acting on A1 receptors. Since, in the presence of DPCPX, there was no significant difference in the vasodilator actions of SNAP during normoxia and hypoxia, it is likely that the disparity seen before DPCPX reflected a greater NO-evoked release of adenosine during hypoxia. Thus, we can propose that the NO donor increased the sensitivity of the endothelial cells to the prevailing level of O2 such that they released adenosine even at normoxic levels of arterial PO2.
Although we have inferred that SNAP caused the release of adenosine from endothelial cells, the possibility must be considered that it caused the additional release of adenosine from skeletal muscle fibres by inhibiting mitochondrial cytochrome oxidase of these fibres (Shen et al. 1995). A previous study indicated that the adenosine released into the interstitial space by skeletal muscle contraction produced vasodilatation by acting on A2A receptors on the vascular smooth muscle (Poucher, 1996), rather than on the A1 receptors that mediate the response to systemic hypoxia (Bryan & Marshall, 1999a) and which the evidence indicates is attributable to endothelial-derived adenosine (see Marshall, 2000). Thus, it seems likely that a large portion of the adenosine released by SNAP was indeed released from the endothelium. In other words, our data are fully consistent with the hypothesis that the release of adenosine from the endothelial cells in systemic hypoxia is the result of a competitive interaction between NO and O2 in endothelial mitochondria.
To test directly whether NO can release adenosine from endothelium, primary porcine aortic endothelial cells were cultured and grown on microcarrier beads and superfused with Krebs-Henseleit solution with a PO2 of ≈ 110 mmHg, which closely resembles that found in arterial blood. In agreement with our hypothesis, both DETA-NO and bradykinin, which are known to cause dilatation by increasing the synthesis of NO, evoked a significant increase in the release of adenosine from endothelial cells. Moreover, whereas the adenosine release evoked by DETA-NO was not sensitive to inhibition of NOS, l-NAME did inhibit the adenosine release evoked by bradykinin. Thus, we have firm evidence that endothelial cells that are under physiological levels of PO2 and releasing NO in response to shear stress (see Clementi et al. 1999), may respond to exogenous or endogenously produced NO by ‘sensing’ hypoxia and releasing adenosine. To our knowledge, this is the first time that NO has been shown directly to cause the release adenosine from endothelial cells. Nevertheless, NO released by a donor was shown to reduce O2 consumption and increase adenosine production in a cGMP-independent manner in quiescent cardiomyocytes, most probably by direct inhibition of the mitochondrial respiratory chain (Stumpe et al. 2001). It therefore seems likely that the ability of NO to release adenosine is a general characteristic of a range of cell types.
In conclusion, we present functional evidence that the presence of local NO is critical for the systemic hypoxia-evoked release of adenosine from the endothelium and, therefore, for hypoxia-evoked vasodilatation in skeletal muscle. The mechanism by which NO achieves this effect is not via its action on soluble guanylate cyclase, but most probably through inhibition of cytochrome oxidase in competition with O2. We propose that in vivo, tonic endothelial synthesis of NO sensitises mitochondrial cytochrome oxidase to decreases in PO2 and that this allows the endothelium to function as a peripheral O2 sensor, releasing adenosine in response to even modest falls in blood O2. Adenosine can then act on smooth muscle receptors to cause dilatation, or on endothelial adenosine receptors to increase NO synthesis and cause vasodilatation. According to our hypothesis, this NO would compete further with O2 at the cytochrome oxidase level, causing further release of adenosine. This positive feedback would be halted when the resulting vasodilatation restores the O2 supply or when maximal vasodilatation is achieved. Further studies are now necessary to establish how this mechanism operates under different physiological and pathological conditions in which NO concentrations are increased by increased shear stress or by agonist stimulation, or when the supply of O2 is diminished.
Acknowledgments
This work was funded by the British Heart Foundation. The authors wish to thank Neale Foxwell for his excellent technical assistance and in growing the PAECs. We also wish to thank Dr R. Campbell for his help with the HPLC analysis and Annie Higgs for her help in the preparation of this manuscript.
Supplementary material
The online version of this paper can be found at:
http://www.jphysiol.org/cgi/content/full/546/2/521
and contains material entitled:
ABP and FBF values recorded during manipulation of NO or cGMP levels after NOS inhibition
The values shown in Tables 1, 2 and 3 are those used to calculate the values of FVC shown in Figs 1, 2 and 3. It may be noted that even when baseline FVC was restored with SNAP infusion or 8-Br-cGMP infusion after L-NAME, ABP did not return to the original baseline level. The values of FBF recorded in each situation can be understood as being the consequence of the changes in FVC and ABP.
References
- Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activation of ATP-sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Metab. 1997;17:100–108. doi: 10.1097/00004647-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Berne RM. The role of adenosine in the regulation of coronary blood flow. Circ Res. 1980;47:807–813. doi: 10.1161/01.res.47.6.807. [DOI] [PubMed] [Google Scholar]
- Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. J Physiol. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeter MWJ, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AHV. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Clementi E, Brown GC, Foxwell N, Moncada S. On the mechanism by which endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci USA. 1999;96:1559–1562. doi: 10.1073/pnas.96.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley KE, Orgway GA, Richardson RS. Deciphering the mysteries of myoglobin in striated muscle. Acta Physiol Scand. 2000;168:623–634. doi: 10.1046/j.1365-201x.2000.00714.x. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle cells isolated from pig coronary artery. J Physiol. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpy E, Costa H, Le Monnier, De Gourville AC. Effects of cyclic GMP elevation on isoprenaline-induced increase on cyclic AMP and relaxation in rat aortic smooth muscle: role of phosphodiesterase 3. Br J Pharmacol. 1996;119:471–478. doi: 10.1111/j.1476-5381.1996.tb15696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deussen A, Moser G, Schrader J. Contribution of coronary endothelial cells to cardiac adenosine production. Eur J Physiol. 1986;406:608–614. doi: 10.1007/BF00584028. [DOI] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. Vasodilatation, oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: roles of nitric oxide. J Physiol. 2001a;532:251–259. doi: 10.1111/j.1469-7793.2001.0251g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. Oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: role of adenosine. J Physiol. 2001b;536:927–935. doi: 10.1111/j.1469-7793.2001.00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryglewski RJ, Moncada S, Palmer RMJ. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF). from porcine aortic endothelial cells. Br J Pharmacol. 1986;87:685–694. doi: 10.1111/j.1476-5381.1986.tb14586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DL, Kessler M, Knauf SK. Regulation of capillary blood flow and oxygen supply in skeletal muscle of dogs during hypoxaemia. J Physiol. 1990;420:431–446. doi: 10.1113/jphysiol.1990.sp017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jöbsis FF. Non-invasive infrared monitoring of cerebral and myocardial oxygen sufficiency and circulation parameters. Science. 1977;198:1264–1267. doi: 10.1126/science.929199. [DOI] [PubMed] [Google Scholar]
- Li J-M, Fenton RA, Wheeler HB, Powell CC, Peyton BD, Cutler BS, Dobson JG. Adenosine A2a receptors increase arterial endothelial cell nitric oxide. J Surg Res. 1998;80:357–364. doi: 10.1006/jsre.1998.5439. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiol Scand. 2000;168:561–573. doi: 10.1046/j.1365-201x.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. The effect of systemic hypoxia on interstitial and blood adenosine, AMP, ADP, ATP in dog skeletal muscle. J Physiol. 2001;536:593–603. doi: 10.1111/j.1469-7793.2001.0593c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhostine N, Lamontagne D. Adenosine contributes to hypoxia-induced vasodilation through ATP-sensitive K+ channel activation. Am J Physiol. 1993;265:H1289–1293. doi: 10.1152/ajpheart.1993.265.4.H1289. [DOI] [PubMed] [Google Scholar]
- Neylon M, Marshall JM. The role of adenosine in the respiratory and cardiovascular response to systemic hypoxia in the rat. J Physiol. 1991;440:529–545. doi: 10.1113/jphysiol.1991.sp018723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JC, Hume AS. In vivo release of cyanide from sodium-nitroprusside. Br J Anaesth. 1987;59:236–239. doi: 10.1093/bja/59.2.236. [DOI] [PubMed] [Google Scholar]
- Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiol Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- Poucher SM. A role of the A2A adenosine receptor subtype in functional hyperaemia in the hind limb of anaesthetised cats. J Physiol. 1996;492:495–503. doi: 10.1113/jphysiol.1996.sp021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray C, Abbas MR, Coney AM, Marshall JM. Interactions of adenosine, prostaglandins and NO in hypoxia-induced vasodilatation: in vivo and in vitro studies. J Physiol. 2002;544:195–210. doi: 10.1113/jphysiol.2002.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal J, La Manna JC, Jöbsis FF, Levasseur JE, Kontos JA, Patterson JL. Effects of respiratory gases on cytochrome A in intact cerebral cortex: is there a critical PO2. Brain Res. 1976;108:143–154. doi: 10.1016/0006-8993(76)90170-0. [DOI] [PubMed] [Google Scholar]
- Schweizer M, Richter C. NO potently and reversibly de-energizes mitochondria at low oxygen tension. Biochem Biophys Res Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- Shen W, Hintze TH, Wolin MS. Nitric oxide: an important signalling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation. 1995;92:1086–1095. doi: 10.1161/01.cir.92.12.3505. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Yudilevich DL, Mann GE. Activation of A2A-purinoceptors by adenosine stimulates l-arginine transport (system y+). and nitric oxide synthesis in human fetal endothelial cells. J Physiol. 1997;499:135–140. doi: 10.1113/jphysiol.1997.sp021916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpe T, Decking UKM, Schrader J. Nitric oxide reduces energy supply by direct action on the respiratory chain in isolated cardiomyocytes. Am J Physiol. 2001;280:H2350–2356. doi: 10.1152/ajpheart.2001.280.5.H2350. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Erecinska M, Brown C, Silver IA. The oxygen dependence of the cellular energy metabolism. Arch Biochem Biophys. 1973;195:485–493. doi: 10.1016/0003-9861(79)90375-8. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Geiger JD, Lautt WW. Improved high-pressure liquid chromatographic-fluorometric assay for measurement of adenosine in plasma. Am J Physiol. 1991;260:G658–664. doi: 10.1152/ajpgi.1991.260.4.G658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.