

Abstract
We previously demonstrated that female mouse ventricles have longer action potential durations (APDs) than males. This delayed repolarization results from a lower current density of the ultrarapid delayed rectifier K+ current (IK,ur) and a lower expression level of its underlying K+ channel (Kv1.5). To evaluate whether this sex difference could be attributable to the action of male sex hormones, we studied the effect of androgen deficiency on ventricular repolarization. We compared cardiac electrophysiological properties in castrated (orchiectomized; ORC) and control (CTL) male mice. Q-Tc intervals as well as APDs measured at 20 %, 50 % and 90 % of repolarization were all significantly longer in ORC than in CTL. The current density of IK,ur was significantly lower in ORC than in CTL (at +50 mV, ORC: 29 ± 4 pA pF−1, n = 25; CTL: 48 ± 5 pA pF−1, n = 17; P = 0.006). In contrast, all the other K+ currents present in mouse ventricular myocytes were comparable between ORC and CTL. Moreover, results of Western blot analysis showed a lower expression level of Kv1.5 protein in ORC but no difference between the two groups for the other K+ channels studied. This study demonstrates that androgen deficiency leads to a reduction in the density of IK,ur and Kv1.5 in mouse ventricle, and consequently, to prolongation of APD and Q-Tc interval. In conclusion, these findings strongly suggest that male sex hormones contribute to the sex difference that we previously reported in cardiac repolarization in adult mouse heart.
Recently, we have shown that there is a sex difference in ventricular repolarization in mouse heart (Trépanier-Boulay et al. 2001). We demonstrated that repolarization was longer in females than in their male counterparts. Indeed, action potential durations (APDs) were significantly longer in myocytes isolated from female ventricle. We then showed that this prolonged repolarization was due to the lower expression of a major repolarizing K+ current, the ultrarapid delayed rectifier K+ current (IK,ur), and of its underlying K+ channel (Kv1.5) in females. In the present study, we investigated one possible mechanism underlying this sex difference.
It is now well recognized that cardiac repolarization is different between men and women (Bazett, 1920; Rautaharju et al. 1992; Lehmann et al. 1997; Yang et al. 1997). As in mice, women have prolonged ventricular repolarization, reflected by a longer rate-corrected Q-T interval (Q-Tc), when compared with men (Rautaharju et al. 1992; Lehmann et al. 1997; Locati et al. 1998; Bidoggia et al. 2000). This sex difference is not observed at birth (Stramba-Badiale et al. 1995) nor during childhood where both girls and boys have long Q-Tc intervals. It is at puberty that the difference appears when boys’ Q-Tc interval shortens (Rautaharju et al. 1992; Lehmann et al. 1997; Locati et al. 1998). Since, at puberty, young men have an important increase in male sex hormones, androgens might be responsible for this Q-T shortening. Recently, Bidoggia et al. (2000) observed that castrated men, who have low levels of androgens, had longer Q-T intervals than ‘intact’ men while virilized women, who have abnormally high levels of male sex hormones, had shorter Q-T intervals than ‘control’ women. The same group also reported that testosterone shortened the Q-T interval in castrated men. Moreover, it has been reported that athletes who take large doses of anabolic androgenic steroids have shorter Q-T intervals (Stolt et al. 1999). Altogether, these studies strongly suggest that androgens may affect repolarization.
Therefore, we carried out the present study to determine whether androgens are involved in the sex difference that we observed in repolarization in mouse heart. The findings presented here clearly show that androgen deficiency induced by castration leads to lengthening of ventricular repolarization in male mouse heart, strongly suggesting a role for male sex hormones in the regulation of cardiac K+ channels.
Methods
Animals
All experiments were performed on 4- to 5-month-old CD1 male mice weighing about 30 g. Castrated (orchiectomized; ORC) and control (CTL) male mice were obtained from Charles River (St-Constant, Québec, Canada). Male mice were castrated at 37 days of age, i.e. just before reaching sexual maturity. ORC mice were always compared with aged-matched CTL males. Sham-operated mice also obtained from Charles River were used for some experiments (see below).
Radioimmunoassay
5α-Dihydrotestosterone (DHT) was measured by radioimmunoassay according to the manufacturer's instructions (Diagnostic Systems Laboratories Inc., TX, USA).
Mouse ventricular myocytes
All experiments conformed with the Canadian Council Animal Care guidelines. Animals were heparinized (100 U, i.p.), anaesthetized by inhalation of isoflurane and then killed by cervical dislocation. Single epicardial myocytes were obtained from the right ventricle of CTL and ORC mice using a previously described cell isolation protocol (Trépanier-Boulay et al. 2001). Briefly, the hearts were rapidly removed, and retrogradely perfused through the aorta on a modified Langendorff apparatus with the following solutions: (i) 5 min with Hepes-buffered Tyrode solution containing (mm): 130 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 0.3 Na2HPO4, 10 Hepes, 5.5 glucose (pH adjusted to 7.4 with NaOH), (ii) 10 min with Tyrode solution without added Ca2+ (‘Ca2+-free’), (iii) 20 min with Ca2+-free Tyrode solution containing 73.7 U ml−1 collagenase Type 2 (Worthington Co. Ltd, Freehold, NJ, USA), 0.1 % bovine serum albumin (BSA; Fraction V, Sigma Chemicals Co., St Louis, Mo, USA), 20 mm taurine and 30 μm CaCl2, and (iv) 5 min with Kraft-Brühe (KB) solution (Isenberg & Klöckner, 1982) containing (mm): 100 potassium glutamate, 10 potassium aspartate, 25 KCl, 10 KH2PO4, 2 MgSO4, 20 taurine, 5 creatine base, 0.5 EGTA, 5 Hepes, 20 glucose and 0.1 % BSA (pH adjusted to 7.2 with KOH).
ECG recordings
Mice were anaesthetized with pentobarbital (65 mg kg−1, i.p.) (Nuyens et al. 2002). Platinum electrodes were placed s.c. and lead I surface ECGs were acquired using the Biopac System MP100 at a rate of 2 kHz. Recordings were analysed using AcqKnowledge 3.7 program (Biopac Systems Inc., Santa Barbara, CA, USA). Mice body temperature was maintained at 37 °C using a heating pad. The Q-T intervals were calculated manually, using a blind-trial procedure, from signal-averaged ECG recordings. The Q-Tc interval was calculated using the formula reported by Mitchell et al. (1998): Q-Tc = Q-T/(R-R/100)1/2.
Electrophysiological recordings
The myocytes were superfused with Hepes-buffered Tyrode solution (see above section Mouse ventricular myocytes). Whole-cell voltage and current recordings were made with a patch-clamp amplifier (Axopatch 200B, Axon Instruments, Foster City, USA). Pipettes were made from borosilicate glass (World Precision Instruments, Sarasota, FL, USA), and had resistances in the range 1.5-4 MΩ when filled with the following solution (mm): 110 potassium aspartate, 20 KCl, 8 NaCl, 1 MgCl2, 1 CaCl2, 10 BAPTA, 4 K2ATP and 10 Hepes (pH 7.2 with KOH). Series resistance (Rs) was between 4 and 8 MΩ, and compensation was applied to reduce Rs by 80-90 %. Voltage-clamp currents were low-pass filtered at 1 kHz with a 4-pole Bessel analog filter, digitized at 4-10 kHz and stored in a microcomputer using pCLAMP 8.0 software (Axon Instruments). All experiments were carried out at room temperature (20-22 °C). K+ currents were recorded in the absence of Na+ or L-type Ca2+ channel blockers to allow recordings of K+ currents and action potentials from the same myocyte. Furthermore, under these recording conditions (e.g. room temperature), ICa is small. Also, the very fast activation and inactivation of the fast sodium current (which represents the largest part of INa; Ju et al. 1996; Nuyens et al. 2002) prevent interference with K+ currents.
K+ current recordings
Current-voltage (I-V) relationships for the total K+ current (Ipeak), for the Ca2+-independent transient outward K+ current (Ito), for IK,ur, for the steady-state outward K+ current (Iss) and for the inwardly rectifying K+ current (IK1) were constructed from the current elicited by a 500 ms voltage-clamp step applied in 10 mV increments from −110 to +50 mV, from a holding potential of −80 mV at a frequency rate of 0.1 Hz. The method of separation of the K+ currents is described in the Results section.
Steady-state inactivation
The voltage dependence of steady-state inactivation for IK,ur was measured using a two-step voltage-clamp protocol consisting of a first 5 s inactivating pulse to selected potentials (between −110 and −20 mV), followed by a second (test) pulse of 2.5 s duration to +30 mV, at a repetition rate of 30 s. In addition, a 100 ms pulse at −40 mV was interposed between the inactivating and test pulses in order to inactivate Ito. IK,ur was obtained by subtraction of the peak test pulse current from the current at the end of the test pulse. The current amplitude of IK,ur at each first pulse potential was normalized to the maximal amplitude of this current (I/Imax), and plotted as a function of the inactivating pre-pulse potential. Data were fitted to a Boltzmann equation:
where V1/2 represents the membrane potential (Vm) at which 50 % of the channels are inactivated and S1/2 is the mid-point slope factor.
Recovery from inactivation
To measure the time- and voltage dependence of recovery from inactivation of IK,ur, a 1.5 s inactivating pulse was followed at intervals between 50 ms and 3 s by a 500 ms test pulse, at a rate of 0.1 Hz. Inactivating and test pulses were both preceded by a brief (100 ms at −40 mV) pulse to inactivate Ito. The holding and interpulse potentials were −80 mV. IK,ur amplitude was measured as the difference between peak outward current and the current 500 ms after the peak. The ratio of current amplitude elicited by the second (test) to the first (inactivating) pulse was plotted as a function of the interpulse interval.
Western blots
Proteins were prepared from mouse hearts (3 pooled ventricles) homogenized in TE buffer (20 mm Tris, 1 mm EDTA, pH 7.4) containing protease inhibitors (leupeptin, aprotinin, benzamidine, PMSF and Na3VO4). The homogenate was centrifuged at 10 000 g. The supernatant was ultracentrifuged 3 times at 200 000 g for 20 min. The pellet was resuspended in TE buffer containing the protease inhibitors and 0.6 m KCl to dissolve contractile proteins. The pellet corresponds to the sarcolemmal-enriched proteins. The Western blot protocols used for analysis of K+ channel protein expression have been reported previously (Trépanier-Boulay et al. 2001).
Confocal imaging
Immunofluorescence analysis and confocal microscopy were carried out on ventricular myocytes isolated from CTL and ORC mice using protocols described previously (Trépanier-Boulay et al. 2001).
Statistical analysis
Results are expressed as means ± s.e.m. Student's unpaired t test was used to compare mean data. The results were considered statistically significant when P values were smaller than 0.05.
Results
Hormone levels
Serum DHT levels were dramatically decreased in ORC compared with CTL male mice ([DHT], ORC: not detectable, n = 5; CTL: 987 ± 246 pg ml−1, n = 4).
Q-T intervals
Figure 1A shows examples of lead I ECG recordings obtained in one CTL and one ORC mouse. A prolonged Q-T interval was observed in the ORC compared with the CTL mouse. As shown in Fig. 1B, Q-Tc intervals were significantly prolonged in ORC compared with CTL male mice. To rule out the possibility that the prolonged cardiac repolarization observed in ORC mice was a direct or indirect consequence of the surgical operation, we recorded surface ECG in sham-operated male mice. As expected, Q-Tc intervals were similar between sham-operated and CTL male mice (sham-operated: 60 ± 4 ms, n = 4; CTL: 66 ± 3 ms, n = 18; P = 0.3).
Figure 1. Comparison of Q-T interval between CTL and ORC mice.
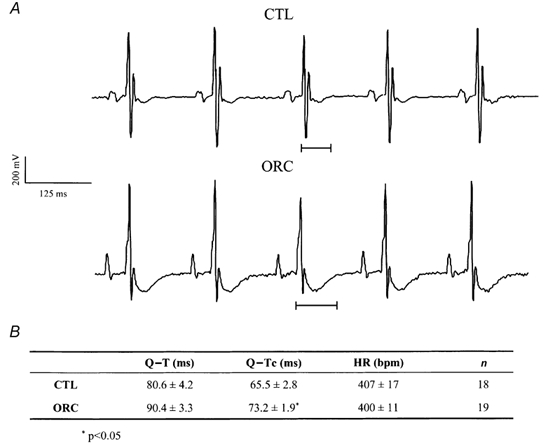
A, examples of lead I surface ECG obtained from one CTL and one ORC male mouse. B, table comparing mean Q-T, Q-Tc and heart rate (HR) in CTL and ORC mice.
Action potential durations
Action potentials were evoked using the whole-cell current-clamp protocol by injection of brief (2-5 ms) stimulus currents (0.4-0.7 nA) at rates of 1 and 4 Hz. Figure 2A shows representative action potentials recorded at 4 Hz in CTL and ORC ventricular myocytes. Figure 2B shows mean APDs measured at 20 %, 50 % and 90 % of repolarization in CTL and ORC myocytes. As for the Q-Tc intervals, the APDs in ORC mice were significantly longer than those in CTL mice (APD20, ORC: 5 ± 0.4 ms; CTL: 3 ± 0.3 ms; P = 0.0004; APD50, ORC: 11 ± 1 ms; CTL: 5 ± 0.4 ms; P = 0.0007; APD90, ORC: 33 ± 3 ms; CTL: 18 ± 2 ms; P = 0.002; n, ORC: 26; CTL:17). Action potentials recorded at 1 Hz were also significantly longer in the ORC group, and this difference was observed for all durations examined (data not shown).
Figure 2. Comparison of action potentials between CTL and ORC mouse ventricular myocytes.
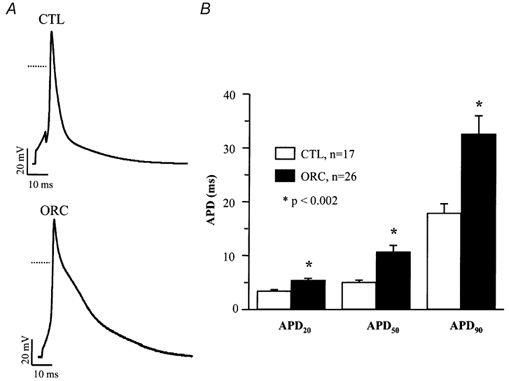
A, typical examples of action potentials recorded from CTL and ORC mice. Dotted lines represent the 0 mV level. B, mean APD at 20 %, 50 % and 90 % of repolarization in CTL and ORC mice. Action potentials were recorded at a frequency of 4 Hz. Recordings shown in this and all subsequent figures were measured at room temperature.
K+ currents
Since K+ currents are major determinants of cardiac repolarization, we compared the K+ currents between CTL and ORC epicardial ventricular myocytes. These included: IK,ur, Ito, Iss and IK1. All currents were normalized to cell capacitance and expressed as densities (pA pF−1). Cell capacitances of ventricular myocytes isolated from ORC and CTL mice were similar (ORC: 82 ± 4 pF, n = 26; CTL: 84 ± 4 pF, n = 24; P = 0.8).
Inward rectifier K+ current: IK1
We compared the current density of IK1 in myocytes obtained from CTL and ORC mice. IK1 was activated by voltage steps ranging from −110 to −40 mV from a holding potential of −80 mV. Figure 3 and Figure 4 show that IK1 was similar in both groups. At −110 mV, the current density was −19 ± 2 pA pF−1 in ORC (n = 26) and −17 ± 2 pA pF−1 in CTL (n = 24; P = 0.2). We also compared the density of IK1 at −60 mV, where this current displays its maximum outward component, and there was no difference between ORC and CTL (1.4 ± 0.2 vs. 1.0 ± 0.2 pA pF−1, P = 0.07). In addition, the resting potential was not significantly different between the two groups (ORC: −74 ± 1 mV; CTL: −73 ± 2 mV, P = 0.5).
Figure 3. Comparison of total K+ current (Ipeak) and IK,slow (IK,ur+Iss) between CTL and ORC mouse ventricular myocytes.
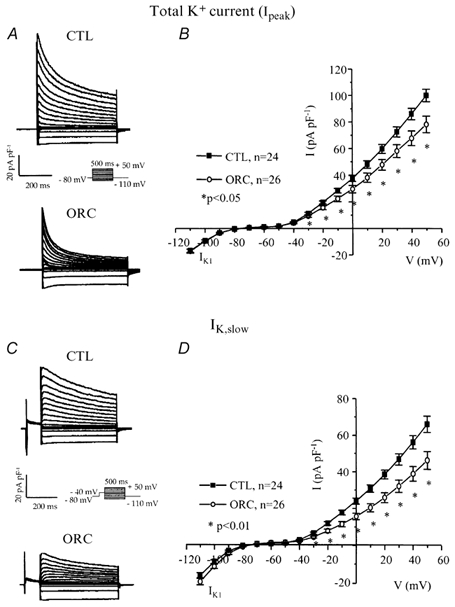
A, family of K+ currents recorded from CTL and ORC myocytes. Membrane currents were activated using the voltage protocol shown in the inset. B, mean I-V relationships for the total K+ current (Ipeak) in CTL and ORC ventricular myocytes. C, superimposed current traces of IK,slow in CTL and ORC cells. IK,slow was activated by 500 ms voltage steps preceded by a 100 ms inactivating prepulse to −40 mV. D, mean I-V curves for IK,slow recorded from CTL and ORC mice. Note that the current densities of IK1, which was activated by voltage steps ranging from −110 mV to −40 mV, were similar between ORC and CTL mice.
Figure 4. Comparison of the transient outward K+ current (Ito) and the steady-state K+ current (Iss) between CTL and ORC mouse ventricular myocytes.
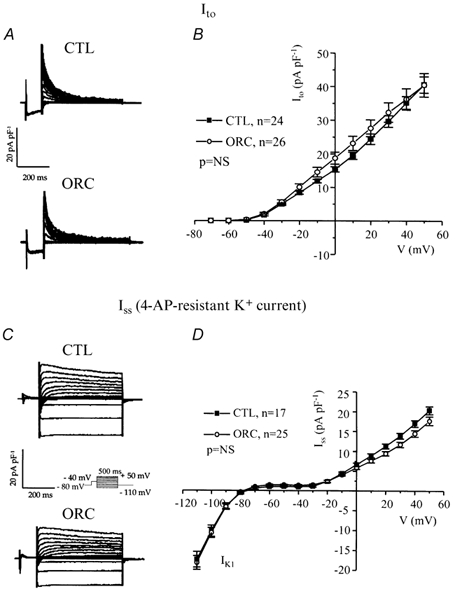
A, superimposed current records illustrating Ito were obtained by subtracting the corresponding currents recorded with (Fig. 3C) and without (Fig. 3A) the inactivating prepulse. B, mean I-V relationships for Ito in CTL and ORC mice. C, representative examples of Iss in CTL and ORC myocytes. Iss was measured after application of 200 μm 4-AP using the inactivation prepulse protocol. D, I-V curves for Iss recorded from CTL and ORC mice. NS, not significant.
Voltage-activated outward K+ currents
Variations in outward K+ current density have dramatic effects on action potential duration (Fiset et al. 1997a). Thus, we examined whether a difference in outward K+ currents between CTL and ORC ventricular myocytes could account for the longer Q-T interval and APD observed in ORC mice. Figure 3A shows a family of K+ currents recorded from ventricular myocytes isolated from CTL and ORC mice. The total K+ current (Ipeak) was activated by a series of test potentials varying from −110 to +50 mV in 10 mV increments from a holding potential of −80 mV. Figure 3B compares mean I-V relationships for Ipeak in CTL and ORC mouse ventricular myocytes. For potentials positive to −40 mV, the density of Ipeak was significantly smaller in ORC cells. For instance, the mean current densities of the peak outward current measured at +50 mV were 78 ± 6 pA pF−1(n = 26) in ORC and 101 ± 5 pA pF−1(n = 24) in CTL (P < 0.01). We then examined the contribution of individual outward K+ currents. First, we eliminated the transient portion (or Ito) by applying an inactivating prepulse (100 ms, −40 mV) immediately before the main activation steps. The current remaining after inactivation of Ito is denoted IK,slow and is composed of IK,ur (or the 4-aminopyridine (4-AP)-sensitive component) and Iss (or the 4-AP-resistant component). Figure 3C shows superimposed current records that correspond to IK,slow in CTL and ORC myocytes. Figure 3D shows the mean I-V plots for IK,slow in CTL and ORC cells where it can be seen that the density of IK,slow was significantly smaller in the ORC group for all potentials positive to −40 mV (at +50 mV, ORC: 46 ± 5 pA pF−1, n = 26; CTL: 66 ± 4 pA pF−1, n = 24; P = 0.004). We then compared the density of Ito, which was obtained by subtracting the current traces measured with and without the inactivating prepulse (in other words, by subtracting IK,slow from Ipeak). Figure 4A shows examples of Ito recorded from CTL and ORC myocytes. As shown in Fig. 4B, there was no difference in the density of Ito between ORC and CTL ventricular myocytes (at +50 mV, ORC: 40 ± 4 pA pF−1, n = 26; CTL: 41 ± 2 pA pF−1, n = 24; P = 0.9). We took advantage of the difference in sensitivity of IK,ur and Iss to the pharmacological agent 4-AP to determine whether the smaller current density of IK,slow was the result of smaller IK,ur and/or Iss in ORC ventricular myocytes. Thus, we applied 200 μm 4-AP (which blocks IK,ur) (Fiset et al. 1997a; London et al. 2001; Trépanier-Boulay et al. 2001) in combination with the inactivating prepulse (which blocks Ito) and recorded the 4-AP-resistant outward K+ current, or Iss (Fig. 4C). It is important to note that 200 μm 4-AP was used to distinguish between IK,ur and Iss rather than IK,ur and Ito. Effectively, as shown above, Ito was measured in the total absence of 4-AP (see Fig. 4A) and this current was always inactivated when 4-AP was used to separate IK,ur and Iss. As can be seen in Fig. 4D, which depicts the mean I-V relationships for Iss, there was no difference between the density of Iss recorded from ORC and CTL mice (at +50 mV, ORC: 18 ± 1 pA pF−1, n = 25; CTL: 20 ± 1 pA pF−1, n = 17; P = 0.08). We then compared the 4-AP-sensitive current (or IK,ur) between CTL and ORC mice. Figure 5A shows superimposed current traces of IK,ur in both groups. These records were obtained by subtracting the currents recorded before (Fig. 3C) and after (Fig. 4C) the addition of 4-AP. As illustrated in these recordings, IK,ur in mouse ventricular myocytes exhibited a much faster inactivation rate than that of human atrial myocytes (Wang et al. 1993; Nygren et al. 1998). The density of IK,ur was markedly smaller in the ORC group than in the CTL group. As shown by the I-V curves presented in Fig. 5B, IK,ur was significantly smaller in the ORC mice over the entire activation range (at +50 mV, ORC: 29 ± 4 pA pF−1, n = 25; CTL: 48 ± 5 pA pF−1, n = 17; P = 0.006).
Figure 5. Comparison of IK,ur between CTL and ORC mouse ventricular myocytes.
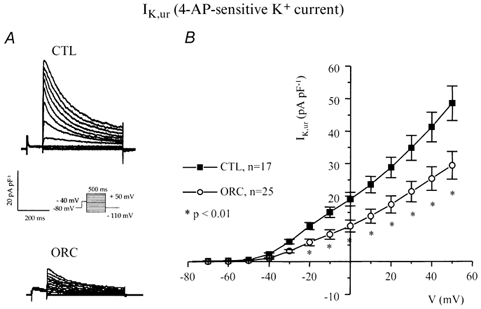
A, family of membrane currents obtained by subtracting pairs of currents recorded with (Fig. 4C) and without (Fig. 3C) application of 200 μm 4-AP in CTL and ORC cells. B, mean I-V curves for IK,ur recorded from CTL and ORC mice.
These voltage-clamp experiments clearly show that the differences we observed in APD and Q-Tc interval between intact and ORC male mice result from the lower current density of IK,ur in the ventricular myocytes isolated from ORC mice.
Voltage dependence of steady-state inactivation of IK,ur
Figure 6A compares the voltage dependence of steady-state inactivation of IK,ur between the two groups. The voltage protocol is shown in the inset. Figure 6B shows Boltzmann functions fitted to mean data recorded in CTL and ORC mice. The voltage dependence of steady-state inactivation of IK,ur was identical in ORC and CTL myocytes (V1/2, ORC: −48 ± 2 mV; CTL: −48 ± 2 mV; P = 0.8; slope factor, ORC: 7 ± 1 mV; CTL: 7 ± 1 mV; P = 0.4; ORC: n = 8; CTL: n = 11).
Figure 6. Comparison of kinetic parameters for IK,ur between CTL and ORC ventricular myocytes.
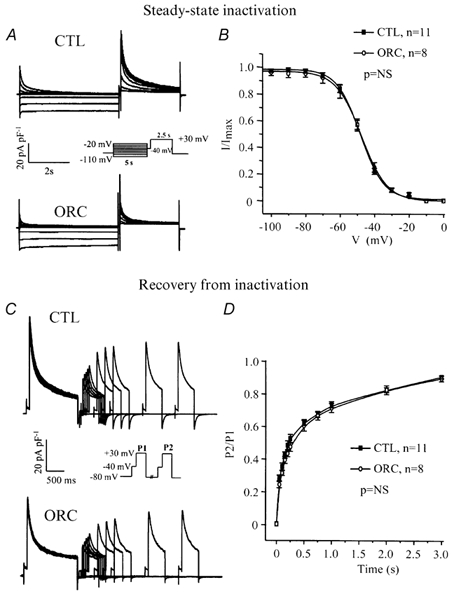
A, superimposed current records showing voltage dependence of steady-state inactivation for IK,ur in CTL and ORC myocytes. The cells were held at various test potentials varying from −110 to −20 mV for 5 s. A 2.5 s voltage step to +30 mV preceded by an inactivating prepulse (at −40 mV for 100 ms) was then applied to measure the remaining current. B, graph comparing the voltage dependence of steady-state inactivation of IK,ur between CTL and ORC mice. I/Imax is the current normalized to the current obtained with the −110 mV voltage step. Smooth lines are best-fit Boltzmann functions. C, family of current recordings showing the time course of recovery from inactivation for IK,ur in CTL and ORC cells. Two voltage steps (+30 mV; P1 = 1500 ms, P2 = 500 ms) separated by 50, 100, 150, 200, 250, 500, 750, 1000, 2000 and 3000 ms were applied. Both steps were preceded by the Ito inactivating prepulse. D, graph comparing reactivation of IK,ur between CTL and ORC myocytes. P2/P1 represents the ratio of the amplitude of the current generated by each pulse.
Recovery from inactivation of IK,ur
Figure 6C shows the results of a voltage-clamp experiment comparing the rate of recovery from inactivation in CTL and ORC mice. As shown in Fig. 6D, ORC and CTL myocytes recovered from inactivation in a similar fashion. The data were best fitted with a single exponential function and mean time constants were 382 ± 45 ms in ORC (n = 8) and 298 ± 39 ms in CTL (n = 11) mice (P = 0.2). These results indicate that alterations in kinetic properties of IK,ur cannot explain the lower density of IK,ur in ORC mice.
Protein expression of K+ channels in CTL and ORC mice
We then examined the expression levels of the following K+ channel isoforms responsible for the currents described above: Kv1.5 (IK,ur; London et al. 2001), Kv2.1 (Iss; Xu et al. 1999), Kv4.2 and Kv4.3 (Ito; Dixon & McKinnon, 1994; Dixon et al. 1996; Fiset et al. 1997b; Barry et al. 1998; Wickenden et al. 1999) and Kir2.1 (IK1; Kubo et al. 1993; Zaritsky et al. 2001). Consistent with the electrophysiological results, data presented in Fig. 7A show that the expression level of Kv1.5 was clearly lower in ORC ventricles than in CTL ventricles. For all the other K+ channels studied, the protein expression was similar between the two groups, as were the current densities of Ito, Iss, and IK1 of ORC and CTL mice. In addition, immunofluorescence and confocal microscopy studies showed a lower expression for Kv1.5 proteins in ORC myocytes (Fig. 7B). This relative reduction of expression was specific for Kv1.5, as exemplified by the result obtained with Kir2.1 (coding for IK1) that showed similar fluorescence intensity between the two groups (data not shown).
Figure 7. Western blot and immunofluorescence detection of K+ channel expression in CTL and ORC male mouse ventricle.
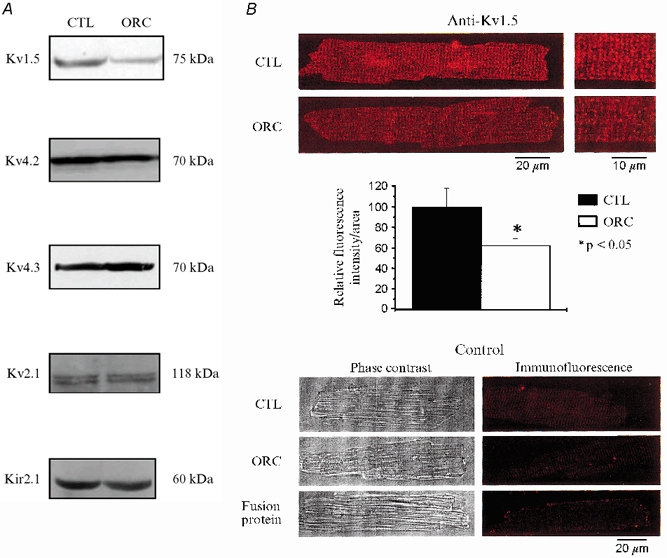
A, comparison of K+ channel protein expression in CTL and ORC ventricles. Western blot analysis of Kv1.5 (1:500), Kv4.2 (1:500), Kv4.3 (1:4000), Kv2.1 (1:300) and Kir2.1 (1:500) in sarcolemmal-enriched proteins (100 μg lane−1) isolated from CTL and ORC mouse ventricles (n = 2 per group; 3 pooled ventricles per n value). Antibodies used were all obtained from Alomone Labs (Jerusalem, Israel), with the exception of Kv1.5, which was purchased from Upstate Biotechnology (Lake Placid, NY, USA). Equal protein loading was confirmed by Ponceau S-stained membranes. Furthermore, we used Kir2.1 as an internal control on the same Western blot gel as Kv1.5 and found no difference in the density of this protein (data not shown). B, immunofluorescence labelling of Kv1.5 in CTL and ORC male mouse ventricular myocytes. Upper panels (left), isolated cells were stained by exposure to the primary antibody and then to TRITC-conjugated donkey anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories Inc., Baltimore, PA, USA). The red fluorescence staining indicates the presence of Kv1.5 in CTL and ORC myocytes. Right, cells seen on the left at higher magnification. Middle panel, bar graph showing the relative fluorescence intensity of Kv1.5 in CTL and ORC myocytes (2 mice per group; 10 cells studied per mouse). Individual values of Kv1.5 fluorescence intensity corresponded to whole-cell fluorescence intensity. These values were obtained with the laser scanning microscopy software using an indicator that recorded fluorescence intensity at every pixel of the cell image. These measures were then normalized to cell surface area to account for cell size. Lower panels, phase-contrast images (left) and immunofluorescence detection (right) of the same CTL and ORC cells. These negative controls show that no staining was apparent when the primary antibody was omitted in CTL and ORC cells. The experiment using a fusion protein specific for the sequence of the antibody shows the specificity of the staining for Kv1.5.
Discussion
We have shown that chronic androgen deficiency alters mouse cardiac repolarization. Males that were subjected to castration exhibited longer Q-Tc intervals and APDs than control mice. This delayed ventricular repolarization was associated with lower current density of IK,ur and lower expression of its corresponding K+ channel, Kv1.5, despite normal voltage dependence and kinetic properties of this current in ORC mice. In contrast, all the other K+ currents/channels were unchanged in ORC mice.
Initially, we examined cardiac repolarization 1-2 months after the castration (as opposed to 3-4 months). At this earlier stage, the current density of IK,ur was also lower in the ORC mice but the reduction did not reach the level of statistical significance (data not shown). As expected, this diminution did not impact on cardiac repolarization since APDs were not significantly different between CTL and ORC mice of 2-3 months of age. Mice less than 1 month old already have considerable amounts of circulating androgens (Overpeck et al. 1978). It is possible that waiting a period of 1-2 months after the castration is not enough to eliminate entirely the effect of endogenous androgens. In addition, it is also likely that long-term androgen deficiency is necessary to affect organs, such as the heart, which are not the primary target of sex hormones (Roy et al. 1997).
The cardiac phenotype of 4- to 5-month-old ORC mice resembles that of female mice (see Trépanier-Boulay et al. 2001 and Table 1). Indeed, compared with males, both females and ORC mice display longer Q-Tc intervals on the ECG. This prolonged repolarization time is associated with a specific decrease in IK,ur density that can be explained by a lower expression of Kv1.5 but not by alterations in the voltage dependence and kinetics of the current. Results presented here strongly suggest that the sex differences observed in mouse cardiac repolarization might be due in part to the action of androgens. Most of the biological effects of sex steroid hormones are mediated through the association with the androgen receptor (Litwack & Schmidt, 1997). The binding of androgen with its receptor results in the formation of an active complex that binds DNA and promotes the transcription of specific genes, giving rise to higher levels of the gene product (Litwack & Schmidt, 1997). Thus, it is possible that androgens would promote Kv1.5 expression and this would result in greater K+ current density, and shorter APD and Q-Tc interval in intact males. Furthermore, consistent with a genomic effect of androgens on this cardiac K+ channel, we have shown that androgen receptors are present in mouse heart (data not shown).
Table 1.
Comparison of Q–T, Q–Tc and heart rate (HR) between male and female mice
| Q–T (ms) | Q–Tc (ms) | HR (bpm) | n | |
|---|---|---|---|---|
| Male | 77.5 ± 3.7 | 64.0 ± 2.3 | 415 ± 14 | 11 |
| Female | 88.7 ± 4.3 | 74.3 ± 2.9* | 427 ± 18 | 11 |
Two-to 3-month-old CD1 mice of both sexes were used. See ECG recordings in Methods.
P < 0.01.
In addition to genomic effects on cardiac repolarization, testosterone can exert direct actions on K+ channels. Indeed, several studies have shown that acute testosterone administration induced vascular relaxation by opening smooth muscle K+ channels (Yue et al. 1995; Chou et al. 1996; Deenadayalu et al. 2001; Ding & Stallone, 2001). While being interesting, this mechanism is probably not responsible for the difference we observed since superfusion of ventricular myocytes with DHT did not affect K+ currents and APD (J. Brouillette & C. Fiset, unpublished observations). In addition, it has been reported that acute perfusion of testosterone did not affect ventricular APDs in the guinea-pig (Jiang et al. 1992) nor Q-T interval in men (White et al. 1999). Also, as mentioned earlier, 1-2 months of androgen deficiency was insufficient to induce changes in cardiac repolarization. The fact that a longer period of time (3-4 months) was required to alter cardiac repolarization in mouse ventricle does not support a non-genomic action of androgens on K+ currents.
Relation to previous studies
Other investigators have studied the effect of chronic DHT treatment on ventricular repolarization. One group observed a decrease in APD in both ovariectomized (OVX) (Hara et al. 1998) and intact female (Pham et al. 2002b) rabbits treated with DHT compared with untreated OVX or female rabbits, respectively. These data support the assumption that androgens shorten ventricular repolarization. However, they did not study K+ currents to demonstrate if this faster repolarization resulted from an increase in K+ currents. On the other hand, Drici et al. (1996) examined K+ channels and found that Kv1.5 and minK were downregulated after 20 day injections of DHT in OVX females. However, the roles of the corresponding K+ currents (IK,ur and the slow component of the delayed rectifier, IK,s) are probably small in the rabbit ventricle.
In contrast to the results presented here, Pham et al. (2001) did not observe any difference in APD between castrated and intact male rabbits. There could be many reasons for this discrepancy. First, rabbits and mice share some but not all of their K+ channels. As mentioned earlier, the physiological role for IK,ur, the K+ current responsible for the longer APD observed in our study, is minor in the rabbit and this could well explain the difference between the two studies. In addition, examination of the experimental design revealed major differences between the two studies. Their rabbits were castrated at an older age (50-60 days vs. 37 days in our study), implying that they were exposed for a longer period of time to androgens, and they were subjected to a shorter period of androgen deficiency (42-49 days vs. a minimum of 90 days in our study). All these factors could explain the absence of effect on cardiac repolarization in their study. In a recent study, the same group showed that Ca2+ currents were not affected by castration nor by DHT replacement in male rabbits (Pham et al. 2002a). That study suggests that modifications in Ca2+ currents between castrated and control males would not be responsible for the difference in cardiac repolarization that we report here. In keeping with this, we have previously reported that there is no sex difference in Ca2+ currents in mouse ventricles (Trépanier-Boulay et al. 2001), suggesting that sex steroid hormones do not alter Ca2+ currents.
Experimental data also suggest that androgens may have a protective role against the antiarrhythmic actions of some cardiotoxic drugs. Shuba et al. (2001) reported that the effects of drugs that inhibit HERG (which codes for the rapid component of the delayed rectifier, IK,r) current in Xenopus oocytes are prevented by pretreatment with testosterone. Moreover, data suggesting a lesser degree of quinidine-induced Q-T prolongation in DHT vs. oestradiol-pretreated ovariectomized rabbits have also been reported (Drici et al. 1996). Finally, a recent study reported that the APD prolongation induced by dofetilide was less important in female rabbits treated with DHT than in control females (Pham et al. 2002b).
The involvement of male sex hormones in the regulation of ventricular repolarization does not rule out a possible role for the female sex hormones in this phenomenon. In fact, 17β-oestradiol has been shown to affect cardiac repolarization in several species including guinea-pig (Nakajima et al. 1999; Tanabe et al. 1999) and rat (Berger et al. 1997). Ongoing studies in our laboratory are focusing on the role of female sex hormones in murine cardiac repolarization.
In conclusion, this work improves our understanding of the role of male sex hormones in the regulation of cardiac K+ channels. The findings presented here strongly suggest that androgens could contribute to sex-based differences in cardiac repolarization. Finally, in addition to displaying sex differences in ventricular repolarization, mouse cardiac K+ currents/channels can be modulated by variations in sex hormone levels, thus reinforcing the validity of mice as an animal model to study sex-related differences in cardiac electrophysiology.
Acknowledgments
This study was supported by operating and personal grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada and Québec, the Research Funds of the Montreal Heart Institute and the Natural Sciences and Engineering Research Council of Canada. We would like to thank Chantale St-Michel, Louis-Robert Villeneuve and Marc-Antoine Gillis for technical assistance.
References
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodelling in mice expressing a dominant-negative Kv4 α-subunit. Circ Res. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Bazett H. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353–370. [Google Scholar]
- Berger F, Borchard U, Hafner D, Pütz I, Weis TM. Effects of 17β-estradiol on action potential and ionic currents in male rat ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:788–796. doi: 10.1007/pl00005119. [DOI] [PubMed] [Google Scholar]
- Bidoggia H, Maciel JP, Capalozza N, Mosca S, Blaksley EJ, Valverde E, Bertran G, Arini P, Biagetti MO, Quinteiro R. Sex difference on the electrocardiographic pattern of cardiac repolarization: possible role of testosterone. Am Heart J. 2000;140:678–683. doi: 10.1067/mhj.2000.109918. [DOI] [PubMed] [Google Scholar]
- Chou TM, Sudhir K, Hutchison SJ, Ko E, Amidon TM, Collins P, Chatterjee K. Testosterone induces dilation of canine coronary conductance and resistance arteries in vivo. Circulation. 1996;94:2614–2619. doi: 10.1161/01.cir.94.10.2614. [DOI] [PubMed] [Google Scholar]
- Deenadayalu VP, White RE, Stallone JN, Gao X, Garcia AJ. Testosterone relaxes coronary arteries by opening the large-conductance, calcium-activated potassium channel. Am J Physiol Heart Circ Physiol. 2001;281:H1720–1727. doi: 10.1152/ajpheart.2001.281.4.H1720. [DOI] [PubMed] [Google Scholar]
- Ding A, Stallone JN. Testosterone-induced relaxation of rat aorta is androgen structure specific and involves K+ channel activation. J Appl Physiol. 2001;91:2742–2750. doi: 10.1152/jappl.2001.91.6.2742. [DOI] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4. 3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL. Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation. 1996;94:1471–1474. doi: 10.1161/01.cir.94.6.1471. [DOI] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Larsen TS, Giles WR. A rapidly activating sustained K+ current modulates repolarization and excitation-contraction coupling in adult mouse ventricle. J Physiol. 1997a;504:557–563. doi: 10.1111/j.1469-7793.1997.557bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Shimoni Y, Giles WR. Shal-type channels contribute to the Ca2+-independent transient outward K+ current in rat ventricle. J Physiol. 1997b;500:51–64. doi: 10.1113/jphysiol.1997.sp021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Danilo P, Jr, Rosen MR. Effects of gonadal steroids on ventricular repolarization and on the response to E4031. J Pharmacol Exp Ther. 1998;285:1068–1072. [PubMed] [Google Scholar]
- Isenberg G, Klöckner U. Calcium tolerant ventricular myocytes prepared by preincubation in a "KB medium". Pflugers Arch. 1982;395:6–18. doi: 10.1007/BF00584963. [DOI] [PubMed] [Google Scholar]
- Jiang C, Poole-Wilson PA, Sarrel PM, Mochizuki S, Collins P, MacLeod KT. Effect of 17β-oestradiol on contraction, Ca2+ current and intracellular free Ca2+ in guinea-pig isolated cardiac myocytes. Br J Pharmacol. 1992;106:739–745. doi: 10.1111/j.1476-5381.1992.tb14403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Lehmann MH, Timothy KW, Frankovich D, Fromm BS, Keating M, Locati EH, Taggard RT, Towbin JA, Moss AJ, Schwartz PJ, Vincent GM. Age-gender influence on the rate-corrected interval and the QT-heart rate relation in families with genotypically characterized long QT syndrome. J Am Coll Cardiol. 1997;29:93–99. doi: 10.1016/s0735-1097(96)00454-8. [DOI] [PubMed] [Google Scholar]
- Litwack G, Schmidt TJ. Biochemistry of hormones II: Steroid hormone. In: Devlin TM, editor. Textbook of Biochemistry with Clinical Correlations. New York: Wiley-Liss; 1997. pp. 893–918. [Google Scholar]
- Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, Towbin JA, Priori SG, Napolitano C, Robinson JL, Andrews M, Timothy K, Hall WJ. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome. Findings from the international LQTS registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- London B, Guo W, Pan X, Lee JS, Shusterman V, Rocco CJ, Logothetis DA, Nerbonne JM, Hill JA. Targeted replacement of Kv1. 5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of IK, slow and resistance to drug-induced QT prolongation. Circ Res. 2001;88:940–946. doi: 10.1161/hh0901.090929. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and QT interval in the conscious mouse. Am J Physiol. 1998;274:H747–751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Iwasawa K, Oonuma H, Morita T, Goto A, Wang Y, Hazama H. Antiarrhythmic effect and its underlying ionic mechanism of 17β-estradiol in cardiac myocytes. Br J Pharmacol. 1999;127:429–440. doi: 10.1038/sj.bjp.0702576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nature Med. 2002;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- Nygren A, Fiset C, Firek JW, Clark JW, Lindblad DS, Clark RB, Giles WR. Mathematical model of an adult human atrial cell. The role of K+ currents in repolarization. Circ Res. 1998;82:63–81. doi: 10.1161/01.res.82.1.63. [DOI] [PubMed] [Google Scholar]
- Overpeck JG, Colson SH, Hohmann JR, Applestine MS, Reilly JF. Concentrations of circulating steroids in normal prepubertal and adult male and female humans, chimpanzees, rhesus monkeys, rats, mice, and hamsters: a literature survey. J Toxic Env Health. 1978;4:785–803. doi: 10.1080/15287397809529700. [DOI] [PubMed] [Google Scholar]
- Pham TV, Robinson RB, Danilo P, Jr, Rosen MR. Effects of gonadal steroids on gender-related differences in transmural dispersion of L-type calcium current. Cardiovasc Res. 2002a;53:752–762. doi: 10.1016/s0008-6363(01)00449-7. [DOI] [PubMed] [Google Scholar]
- Pham TV, Sosunov EA, Anyukhovsky EP, Danilo P, Jr, Rosen MR. Testosterone diminishes the proarrythmic effects of dofetilide in normal female rabbits. Circulation. 2002b;106:2132–2136. doi: 10.1161/01.cir.0000033596.21845.d8. [DOI] [PubMed] [Google Scholar]
- Pham TV, Sosunov EA, Gainullin RZ, Danilo P, Jr, Rosen MR. Impact of sex and gonadal steroids on prolongation of ventricular repolarization and arrhythmias induced by IK-blocking drugs. Circulation. 2001;103:2207–2212. doi: 10.1161/01.cir.103.17.2207. [DOI] [PubMed] [Google Scholar]
- Rautaharju PM, Zhou SH, Wong S, Calhoun HP, Berenson GS, Prineas R, Davignon A. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol. 1992;8:690–695. [PubMed] [Google Scholar]
- Roy AK, Vellanoweth RL, Jung MH, Chatterjee B. Cellular and molecular effects of androgenic-anabolic steroids. In: Thomas JA, Colby HD, editors. Endocrine Toxicology. UK: Taylor & Francis; 1997. pp. 213–225. [Google Scholar]
- Shuba YM, Degtiar VE, Osipenko VN, Naidenov VG, Woosley RL. Testosterone-mediated modulation of HERG blockade by proarrhythmic agents. Biochem Pharmacol. 2001;62:41–49. doi: 10.1016/s0006-2952(01)00611-6. [DOI] [PubMed] [Google Scholar]
- Stolt A, Karila T, Viitasalo M, Mäntysaari M, Kujala UM, Karjalainen J. QT interval and QT dispersion in endurance athletes and in power athletes using large doses of anabolic steroids. Am J Cardiol. 1999;84:364–366. doi: 10.1016/s0002-9149(99)00299-4. [DOI] [PubMed] [Google Scholar]
- Stramba-Badiale M, Spagnolo D, Bosi G, Schwartz PJ. Are gender differences in QTc present at birth? Am J Cardiol. 1995;75:1277–1278. [PubMed] [Google Scholar]
- Trépanier-Boulay V, St-Michel C, Tremblay A, Fiset C. Gender-based differences in cardiac repolarization in mouse ventricle. Circ Res. 2001;89:437–444. doi: 10.1161/hh1701.095644. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- White C, Ferraro-Borgida M, Moyna N, Mcgill C, Ahlberg A, Thompson P, Heller G. The effect of pharmacokinetically guided acute intravenous testosterone administration on electrocardiographic and blood pressure variables. J Clin Pharmacol. 1999;39:1038–1043. doi: 10.1177/00912709922011809. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Lee P, Sah R, Huang Q, Fishman GI, Backx PH. Targeted expression of a dominant-negative Kv4. 2 K+ channel subunit in the mouse heart. Circ Res. 1999;85:1067–1076. doi: 10.1161/01.res.85.11.1067. [DOI] [PubMed] [Google Scholar]
- Xu H, Barry DM, Li H, Brunet S, Guo W, Nerbonne JM. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 α subunit. Circ Res. 1999;85:623–633. doi: 10.1161/01.res.85.7.623. [DOI] [PubMed] [Google Scholar]
- Yang H, Elko PFBS, Baga JJ, Pires AA, Schuger CD, Steinman RT, Lehmann MH. Maximal ascending and descending slopes of the T wave in men and women. J Electrocardiol. 1997;30:267–276. doi: 10.1016/s0022-0736(97)80038-6. [DOI] [PubMed] [Google Scholar]
- Yue P, Chatterjee K, Beale C, Poole-Wilson PA, Collins P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation. 1995;91:1154–1160. doi: 10.1161/01.cir.91.4.1154. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2. 1 and Kir2.2 genes. J Physiol. 2001;533:697–710. doi: 10.1111/j.1469-7793.2001.t01-1-00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]