

Abstract
Whole-cell recording methods and fluorescence microscopy were used to study the effects of acute exposure to thyroid hormone (T3) on cat atrial myocytes. Acute exposure (≈5 min) to 10 nm T3 significantly increased tetrodotoxin (TTX)-sensitive inward Na+ current (INa) at voltages between −40 and +20 mV. At maximal INa activation (−40 mV) T3 increased peak INa by 32 %. T3 had no effect on the time course of INa decay, voltage dependence of activation, inactivation, or recovery from inactivation. Comparable exposures to reverse T3 (rT3) or T4 had no effect on INa. L-type Ca2+ current was unaffected by acute exposure to T3. T3-induced increases in INa were unaffected by 50 μm nickel, a blocker of T-type Ca2+ current. T3 significantly increased cell shortening (+62 %) and could elicit spontaneous action potentials arising from Ca2+-mediated after-depolarizations. T3 (but not rT3) significantly increased baseline intracellular Ca2+, release of Ca2+ from sarcoplasmic reticulum (SR) and caffeine (10 mm)-induced release of SR Ca2+. We conclude that acute T3 exposure increases Na+ influx via INa and thereby stimulates reverse-mode Na+-Ca2+ exchange to increase intracellular Ca2+ content and release. As a result, T3 increases contraction strength, and can initiate Ca2+-mediated arrhythmic activity. Acute non-genomic effects of T3 can contribute to the positive inotropy and sinus (atrial) tachycardia traditionally attributed to chronic, genomic effects of elevated thyroid hormone on atrial muscle.
Clinically, patients with hyperthyroidism experience elevated heart rate and cardiac contractility with increased stroke volume and cardiac output (Klein & Levey, 2000; Klein & Ojamaa, 2001). Sinus tachycardia at rest and during exercise is the most consistent rhythm disturbance in patients with thyrotoxicosis, with 5-10 % of patients exhibiting atrial fibrillation (Klein & Levey, 2000). The chronic effects of triiodothyronine (T3), the biologically active form of thyroid hormone, are mediated via nuclear receptors that allow the hormone-dependent regulation of transcription (Brent, 1994). The phenotypic changes that result from T3 action on the heart enhance cardiomyocyte contractile function and ion channel properties (Freedberg et al. 1970; Klein & Ojamaa, 2001). In addition, hyperthyroidism enhances sarcoplasmic reticulum (SR) Ca2+ uptake and release by increasing protein content of SR Ca2+-activated ATPase (SERCA2) and the ryanodine receptor while decreasing phospholamban (Kiss et al. 1995; Shenoy et al. 2001). Chronic T3 exposure also increases slow inward Ca2+ current (Binah et al. 1987; Rubinstein & Binah, 1989), outward rectifying K+ current (IK) (Rubinstein & Binah, 1989) and transient outward K+ current (Shimoni & Severson, 1996; Sun et al. 2000).
Acute exposure to T3 also is cardioactive via non-genomic mechanisms of action (Davis & Davis, 1993). T3 receptors are present in cardiac plasma membranes (Segal, 1990). Acute T3 exposure slows inactivation of fast Na+ current in neonatal cardiac myocytes (Harris et al. 1991), increases single channel Na+ current in rat ventricular myocytes (Dudley & Baumgarten, 1993), and increases cellular Ca2+ uptake in rat heart slices (Segal, 1990) and isolated perfused heart (Gotzsche, 1994). Although it is well known that hyperthyroid states exert profound effects on atrial function, the vast majority of research has been performed on ventricular muscle. Consequently, less is known about the acute effects of T3 on atrial myocytes. The purpose of the present study, therefore, was to determine whether acute exposure to T3 exerts non-genomic effects on atrial myocytes that may contribute to the effects of thyroid hormone traditionally attributed to chronic, genomic mechanisms. The present results indicate that acute exposure of atrial myocytes to T3 increases INa, resulting in elevation of intracellular Ca2+ mediated by Na+-Ca2+ exchange. As a result, T3 exerts positive inotropic effects and can contribute to the development of Ca2+-mediated atrial tachy-dysrhythmias.
Methods
Methods used to isolate atrial myocytes from cat heart have been published in detail previously (Wang & Lipsius, 1995). Adult cats of either sex were anaesthetized with sodium pentobarbital (50 mg kg−1, intraperitoneal). Following a bilateral thoracotomy, the heart was rapidly excised and attached to a Langendorff perfusion apparatus. Hearts were perfused with enzyme (0.06 % collagenase; Worthington Biochemical, type II) to disperse individual cells. Cell isolation was performed on the morning of each experiment.
Cells used for study were transferred to a small (0.4 ml) tissue bath on the stage of an inverted microscope. Cells selected for study were elongated and quiescent, and superfused at ≈5 ml min−1 with a modified Tyrode solution containing (mm): NaCl 137, KCl 5.4, MgCl2 1, CaCl2 2, Hepes 5, glucose 11, titrated with NaOH (pH of 7.4). Inward Na+ current (INa) was recorded in a whole-cell ruptured patch recording configuration at room temperature. The internal pipette solution contained (mm): caesium glutamate 100, CsCl 40, MgCl2 1, Na2-ATP 4, EGTA 10, Hepes 10, titrated with CsOH to a pH of 7.2. In the ruptured patch method, the junction potential (10 mV) measured between the internal pipette and bath solutions was subtracted from all voltage measurements. To better control INa, external Na+ concentration was reduced by replacement with equimolar TEA-Cl. The external recording solution contained (mm): NaCl 50, TEA-Cl 67, KCl 5.4, MgCl2 1, CaCl2 2, 20 mm CsCl, Hepes 5, glucose 11, titrated with CsOH to pH 7.35. In addition, INa was isolated by adding 5 m verapamil to the external solutions, to block L-type Ca2+ current (ICa,L), and 2 mm 4-aminopyridine, to block transient outward currents. Even with these modifications, it was not possible to completely control INa. Nevertheless, membrane voltage in response to the command pulse did not change between control and test (T3) and therefore the effects of T3 on peak INa cannot be attributed to changes in clamp voltage. ICa,L, action potentials and cell shortening were recorded by a perforated patch (nystatin) method (Horn & Marty, 1988). Nystatin was dissolved in DMSO at a concentration of 50 mg ml−1, and then added to the internal pipette solution to yield a final nystatin concentration of 150 μg ml−1. When recording ICa,L in the perforated patch configuration the internal pipette solution contained (mm): caesium glutamate 100, CsCl 40, MgCl2 1, Na2-ATP 4, EGTA 0.5, Hepes 10, titrated with CsOH to a pH of 7.2. When recording action potentials in the perforated patch configuration the internal pipette solution contained (mm): potassium glutamate 100, MgCl2 1, Na2-ATP 4, KCl 40, EGTA 0.5, Hepes 10, titrated with KOH to pH 7.2. Peak ICa,L amplitude was measured with respect to steady-state current and was not compensated for leak currents. Action potentials were elicited by 3 ms pulses and unloaded cell shortening was measured using a video-based edge detector (Crescent Electronics). A single suction pipette was used to record action potential (bridge mode) and ionic currents (discontinuous voltage clamp mode) using an Axoclamp 2A amplifier (Axon Instruments, Inc., Foster City, CA, USA). The discontinuous (switch clamp) mode precludes the need to compensate for series resistance. In the voltage clamp mode the amplifier sampling rate was approximately 10-12 kHz. A second scope was used to monitor the duty cycle to ensure complete settling of the voltage transient between samples. Computer software (pCLAMP 8; Axon Instruments, Inc.) was used to deliver voltage protocols, acquire and analyse data.
In general, INa was activated by depolarizing voltage clamp steps (80 ms) from a holding potential of −80 mV in 10 mV increments. Peak INa was measured with respect to steady-state current and was not compensated for leak currents. The concentration of 10 nm T3 used in the present study is consistent with serum total T3 levels found in hyperthyroid patients (Chopra & Sabatino, 2000). Cells were exposed to T3 until its effects on INa stabilized, which usually occurred by approximately 5 min. After establishing the ruptured patch configuration, INa amplitude exhibited a ‘run down’ of about 10 % (44 ± 5 to 40 ± 4 pA pF−1; n = 10) that reached a relatively stable level within approximately 5 min. Experiments were begun after this time. Current density (pA pF−1) was obtained by normalizing total current-to-cell capacitance.
Voltage dependence of INa activation was determined by measuring tail currents as follows: an atrial cell was depolarized from −80 mV and clamped for approximately 2 ms, or a duration not leading to channel inactivation, in 10 mV steps from −80 to +50 mV. After the conditioning prepulse to various voltages, the cell was clamped back to −60 mV and the tail currents elicited were measured. Steady-state activation was determined by comparing the tail currents elicited at each varying prepulse voltage with the maximum current elicited, yielding a normalized activation curve. The same protocol was performed in the absence and presence of T3.
Voltage dependence of steady-state inactivation of INa was determined by first applying a depolarizing voltage step to maximally activate INa. Then, the cell was held at −120 mV and clamped in 10 mV increments to +20 mV for 1 s. Following this initial conditioning pulse, the cell was returned to −120 mV to 2 ms, after which the cell was clamped to −40 mV to activate INa. Inward currents, elicited by the test pulse to −40 mV, were normalized to that obtained in the absence of a preceding conditioning pulse and compared in the absence and presence of T3. Data were normalized and fitted using a Boltzmann function: It/Imax = {1 + exp[(Vc – V0.5)/k]}−1, where Vc is the voltage of the conditioning pulse, V0.5 is half-maximal activation or inactivation voltage and k is the slope factor.
The curve for recovery from inactivation was determined by holding the cell at −110 mV and clamping to −40 mV for 1 s. This initial conditioning pulse was followed by a variable time interval (from 0.2 ms to 150 ms) at −110 mV after which the cell was test clamped again to −40 mV for 50 ms. Test current was compared with the conditioning current (Itest/Icond) at each recovery time to yield a normalized value.
Intracellular Ca2+ concentration ([Ca2+]i) was measured using the fluorescent Ca2+ indicator indo-1, as previously described (Hüser et al. 2000b). Myocytes were loaded with Ca2+ indicator by exposure to 5 μm acetoxymethyl ester of indo-1 (indo-1 AM, Molecular Probes, Eugene, OR, USA) in 1 ml Tyrode solution containing 0.001 g ml−1 of a Pluronic F-127 for 10 min at room temperature. Cells were then washed for 10 min to allow de-esterification of the indicator. A coverslip with the attached cells was mounted on the stage of an inverted microscope. For spatially averaged single cell [Ca2+]i measurements indo-1 fluorescence was excited at 357 nm. Cellular fluorescence signals were recorded simultaneously at 405 nm (F405) and 485 nm (F485). Changes of [Ca2+]i are expressed as changes in the ratio R = F405/F485. [Ca2+]i transients were evoked by field stimulation using 3 ms duration suprathreshold voltage pulses delivered through a pair of extracellular platinum electrodes. [Ca2+]i transient amplitude was measured as the difference between baseline and peak [Ca2+]i. Cells were electrically stimulated at 1 Hz and experiments were performed at room temperature.
Data are expressed as mean ± s.e.m. and were statistically analysed for significance using paired and unpaired Student's t test at P < 0.05. The n value indicates the number of cells tested. Drugs and chemicals included: triiodo-l-thyronine (T3); reverse T3 (rT3); l-thyroxine (T4); ryanodine; verapamil; 4-aminopyridine (all from Sigma); tetrodotoxin (TTX) (Calbiochem). The procedures used in this study were in accordance with the guidelines of the Animal Care and Use Committee of Loyola University Medical Centre.
Results
Figure 1A shows superimposed traces of INa elicited by depolarizing clamp steps from −80 to −40 mV in the absence and presence of 10 nm T3. Exposure to T3 for approximately 5 min increased INa from −17 to −26 pA pF−1. There were no discernable effects of T3 on holding current or the time course of INa decay. Washout of T3 returned INa to −20 pA pF−1. In a total of seven cells studied, acute exposure to T3 increased INa from −34 ± 7 to −45 ± 10 pA pF−1 (+32 %; P < 0.05). Washout of T3 for approximately 5 min returned INa to −34 ± 7 pA pF−1. Panel B shows the effects of T3 on the INa current-voltage (I-V) relationship. T3 significantly increased INa at voltages from −40 to +20 mV without affecting the threshold of INa activation, the voltage at which peak INa was activated (-40 mV) or the reversal potential (n = 7). Although the effect of T3 to increase INa was quite small at more positive voltages, paired statistical analysis indicated a significant difference between control and test conditions. Further experiments showed that acute exposure to 10 nm reverse T3 (rT3) or 10 nm T4 had no effect on INa compared with control values throughout the voltage range of −80 to +60 mV (n = 3; data not shown).
Figure 1. Acute exposure (5 min) to 10 nm T3 increases INa.
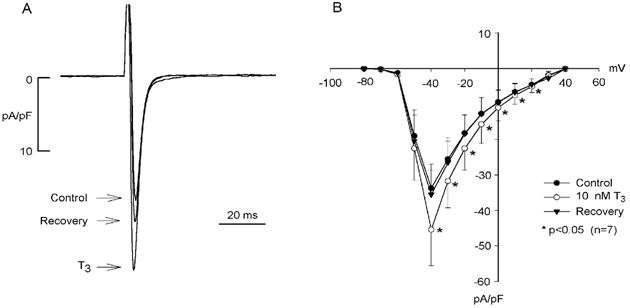
A, INa was elicited by depolarizing clamp steps from a holding potential of −80 mV to −40 mV. T3 elicited a reversible increase in peak INa (+17 %). B, the current-voltage relationship shows that T3 significantly increased INa at voltages between −40 and +20 mV without affecting peak voltage dependence of INa activation or reversal potential. The effects of T3 were fully reversible.
Figure 2 shows that the inward current increased by 10 nm T3 (+23 %) is completely blocked by 10 μm tetrodotoxin (TTX). A total of five cells showed similar results. Additionally, when the holding potential was shifted to −40 mV, a voltage which inactivates INa, depolarizing steps (in the presence of TTX) failed to elicit any inward current (data not shown). Together, these findings indicate that T3 increases TTX-sensitive INa, and that the effect is specific to T3.
Figure 2. Tetrodotoxin (TTX) blocks the inward current increased by T3.
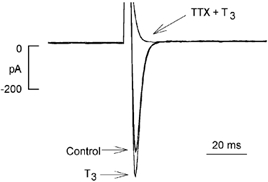
Acute exposure to 10 nm T3 increased the inward current activated by depolarization from −80 to −40 mV. Exposure to 10 μm TTX in the presence of T3 blocked all inward current.
Further experiments showed that the effect of T3 to increase INa is not due to alterations in Na+ channel kinetics. Acute exposure to 10 nm T3 had no effect on the time course of INa decay (inactivation) (see Fig. 1A and Fig. 2). Moreover, the steady-state voltage of half-inactivation was not different in the absence (-83.5 mV) or presence (-82.5 mV) of T3(n = 10). The Boltzmann slope factors (k = 7) also were the same under both conditions. The threshold of activation (-60 mV) and maximum INa activation (-20 mV) were unchanged by T3. Also, INa reached 90 % recovery from inactivation in approximately 25 ms under both conditions.
Chronic exposure to thyroid hormone increases ICa,L (Binah et al. 1987). We therefore examined the acute effects of T3 on ICa,L. Figure 3 shows selected recordings of ICa,L in the absence (A) and presence (B) of 10 nm T3 and the effect of T3 on the I-V relationship of ICa,L (C). Activation of ICa,L from a holding potential of −40 mV inactivates Na+ channels and therefore eliminates any potential secondary effects of T3 mediated via stimulation of INa. Acute exposure to T3 (for up to 15 min) had no effect on ICa,L throughout the voltage range of activation (n = 5). At peak activation voltage (0 mV), ICa,L amplitude was not significantly different in the absence (-461 ± 54 pA) and presence (-468 ± 63 pA) of T3.
Figure 3. Acute exposure to 10 nm T3 has no effect on L-type Ca2+ current (ICa,L).
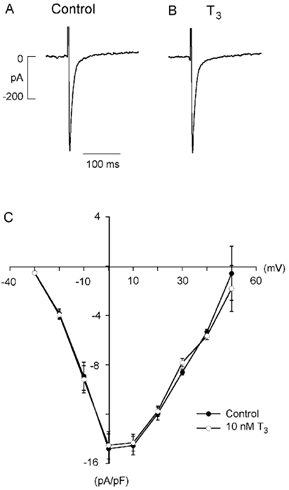
A, control ICa,L elicited by depolarizing clamp steps from −40 to 0 mV. B, T3 had no effect on peak ICa,L or ICa,L inactivation. C, current-voltage relationship shows T3 had no effect on voltage dependence of peak ICa,L activation or reversal potential.
The relatively negative voltage range of INa activation overlaps with the activation range of T-type Ca2+ current (ICa,T). We therefore examined whether the effects of T3 to increase inward current could be mediated, in part, via ICa,T by testing the effects of T3 in the absence and presence of 50 μm nickel, an effective blocker of ICa,T (Zhou & Lipsius, 1994). Maximal INa was activated by voltage steps from −80 to −40 mV. In a total of five cells tested, acute exposure to 10 nm T3 elicited a typical increase in peak INa from a control value of −46 ± 2 to −55 ± 4 pA pF−1. Addition of 50 μm nickel had no significant effect (-53 ± 3 pA pF−1) on T3-induced increases in INa (data not shown), suggesting that ICa,T was not responsible for the increase in inward current induced by T3.
Stimulation of Na+ influx via INa can stimulate Ca2+ uptake via reverse-mode Na+-Ca2+ exchange and thereby load SR Ca2+ stores (Sipido et al. 1995). By this mechanism, T3 would be expected to enhance cell shortening. To minimize intracellular dialysis and avoid buffering of intracellular Ca2+ we used a perforated patch recording method to record action potentials and trigger contractions. As shown in Fig. 4, action potentials (top) and cell shortening (bottom) were elicited by electrical stimulation at 1 Hz. Although acute exposure (5 min) to 10 nm T3 did not significantly affect action potential configuration (action potential duration at 90 % repolarization: control; 353 ± 5 vs. T3; 350 ± 12 ms; n = 4), T3 prominently increased cell shortening (63 ± 15 %; n = 4). Note that the positive inotropic effects of T3 were reversed within minutes of removing T3 from the bath.
Figure 4. Acute exposure to 10 nm T3 increases action potential-induced contraction.
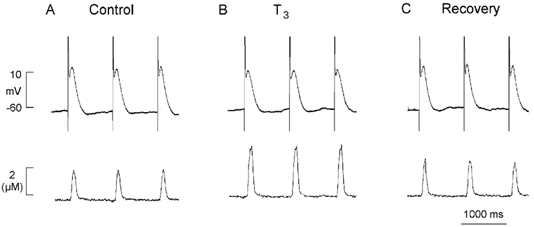
A, control recording of atrial action potentials and contraction elicited by field stimulation at 1 Hz. B, T3 had little effect on action potential configuration but significantly increased contraction amplitude. C, washout of T3 from the tissue bath returned contraction amplitude toward control (recovery).
Acute exposure to T3 also could elicit spontaneous activity arising from delayed after-depolarizations. Figure 5A shows a typical response where acute exposure of a stimulated (1 Hz) atrial cell to 10 nm T3 led to the development of delayed after-depolarizations and spontaneous action potentials (asterisks). Similar results were obtained in 11 of 15 cells tested. Withdrawing T3 abolished abnormal activity and restored control conditions. Figure 5B shows that after-depolarizations and spontaneous beats elicited by T3 were abolished by exposure (2 min) to 10 μm ryanodine (Rya), an alkaloid that interferes with SR Ca2+ release. Similar results were obtained in a total of four cells studied. During exposure to T3, stimulated action potential durations were variable, depending on whether the stimulated action potential was preceded by a spontaneous Ca2+-mediated depolarization. This is consistent with the idea that the configuration of each stimulated action potential was modulated by the amount of SR Ca2+ available for release and its effect of stimulating Na+-Ca2+ exchange current. The fact that ryanodine normalized action potential duration in the presence of T3 supports this interpretation. These findings indicate that T3 can induce overload of SR Ca2+ content, presumably by enhancing Na+ influx via INa.
Figure 5. Acute exposure to 10 nm T3 can induce delayed after-depolarizations and spontaneous activity.
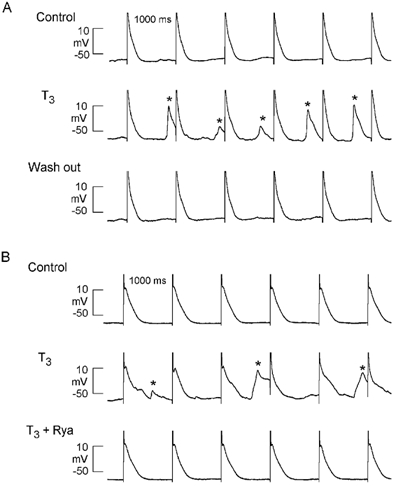
A, control recordings show action potential elicited by field stimulation at 1 Hz; T3 induced delayed after-depolarizations that reached threshold to form spontaneous (non-driven) action potentials (*); washout of T3 abolished abnormal activities and returned action potential configuration to control. B, control action potentials elicited by field stimulation at 1 Hz; T3 induced delayed after-depolarizations and spontaneous beats (*); addition of 10 μm ryanodine (Rya) abolished abnormal activities and returned action potential configuration to control.
In the following experiments we used the Ca2+-sensitive fluorescent indicator, indo-1, to visualize the effects of T3 on [Ca2+]i. Figure 6A shows control [Ca2+]i transients elicited by field stimulation at 1 Hz recorded from the same cell. Exposure to 10 nm T3 for 5 min increased both baseline intracellular Ca2+ and peak Ca2+ transient amplitudes. After 10 min of exposure, the effects of T3 reached steady-state with only small further increases in both baseline [Ca2+]i and peak [Ca2+]i transient amplitude. Note that within minutes of washing out T3, baseline [Ca2+]i and [Ca2+]i transient amplitude returned to control levels. Similar experiments showed that 10 nm rT3 had no effect on [Ca2+]i (n = 4; data not shown). In a total of seven cells, at 5 min of exposure, T3 significantly increased baseline [Ca2+]i (control, 2.03 ± 0.05 vs. T3, 2.32 ± 0.06; +14 %; P < 0.05) and [Ca2+]i transient amplitude (control, 1.13 ± 0.13 vs. T3, 2.11 ± 0.2; +87 %; P < 0.05). At 10 min of T3 exposure, further increases in baseline [Ca2+]i and [Ca2+]i transient amplitude were not statistically significant, indicating that steady-state effects of T3 were reached.
Figure 6. Acute exposure to 10 nm T3 increases intracellular Ca2+ content and release.
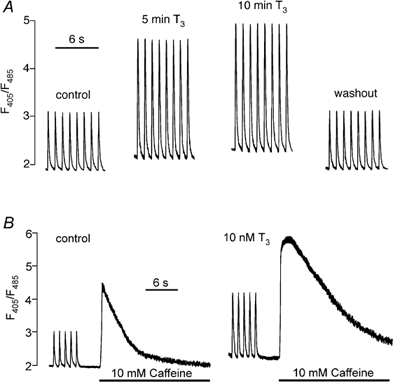
A, control [Ca2+]i transients elicited by field stimulation at 1 Hz. Exposure to T3 for 5 min increased baseline [Ca2+]i and [Ca2+]i transient amplitude without affecting relaxation of [Ca2+]i transients. After 10 min of exposure to T3 there were further increases in both baseline [Ca2+]i and [Ca2+]i transient amplitude. Washout of T3 restored control parameters. B, control recordings from another atrial cell show [Ca2+]i transients followed by SR Ca2+ release induced by rapid exposure to10 mm caffeine. Acute exposure to 10 nm T3 increased baseline [Ca2+]i, [Ca2+]i transient amplitude and SR Ca2+ release induced by 10 mm caffeine.
To determine whether T3 increased SR Ca2+ content, single atrial myocytes were rapidly exposed to 10 mm caffeine to release SR Ca2+. Caffeine was tested in the same cell before (control) and during a 5 min exposure to 10 nm T3. Cells were field-stimulated at 1 Hz for approximately 5 min before each caffeine exposure to ensure steady-state loading of SR Ca2+. The left trace of Fig. 6B shows control steady-state [Ca2+]i transients elicited by field stimulation. Within approximately 5 s of switching off the stimulation, rapid exposure to 10 mm caffeine elicited a large [Ca2+]i transient. The right trace of Fig. 6B shows that a 5 min exposure to T3 increased the baseline [Ca2+]i, the amplitude of the [Ca2+]i transients and the caffeine-induced [Ca2+]i transient compared with control. In a total of three cells, acute exposure to T3 significantly increased the caffeine-induced [Ca2+]i transient compared with control (control, 2.49 ± 0.05 vs. T3, 3.39 ± 0.14; +36 %; P < 0.05). Together, these findings indicate that acute T3 exposure increases intracellular SR Ca2+ content and release.
Discussion
The present results demonstrate that in atrial myocytes, acute exposure to T3 increases TTX-sensitive INa. As a result of enhanced Na+ influx, T3 indirectly increases intracellular Ca2+ via Na+-Ca2+ exchange, leading to positive inotropic effects and possible Ca2+-mediated dysrhythmias. The acute effects of T3 on INa were achieved within 5 min of exposure, a time frame too short to be consistent with genomic effects. This relatively rapid response to T3 is consistent with other studies in which acute exposure to T3 elicited rapid membrane-mediated changes in transmembrane transport of ions and substrates (Segal, 1989). Moreover, as shown in the present study, the stimulatory effects of T3 on INa, cell shortening, the development of arrhythmic activities and [Ca2+]i transients were all reversible within minutes of removing T3 from the tissue bath. Together, these findings support the idea that in the present experiments acute exposure to T3 acted via non-genomic mechanisms.
T3-induced increases in macroscopic INa in atrial myocytes are consistent with the effect of T3 to increase single Na+ channel bursting behaviour in ventricular myocytes (Dudley & Baumgarten, 1993). In the present experiments, T3 acutely increased INa by about 32 % within approximately 5 min. However, as a result of cell dialysis, ‘run down’ decreased INa by approximately 10 % over the same time period. Moreover, the effects of T3 on INa were studied at room temperature and in one-third the normal extracellular Na+ concentration. Each of these factors would be expected to attenuate the absolute effects of T3 on INa. It therefore seems likely that these experiments underestimate the actual stimulatory effect of T3 on INa amplitude. This idea is supported by the present findings that T3 more than doubled the stimulated [Ca2+]i transient amplitude and cell shortening. Several additional factors may contribute to these prominent effects of T3 on [Ca2+]i. First, the effects of T3 on [Ca2+]i transients and cell shortening were tested in cells stimulated by action potentials in normal extracellular Na+ concentration. In addition, Ca2+ influx via reverse-mode Na+-Ca2+ exchange, especially following Na+ influx via INa, can contribute directly to triggering SR Ca2+ release (Leblanc & Hume, 1990; Lipp & Niggli, 1994; Lipp et al. 2002). Furthermore, an increase in SR Ca2+ content can increase the fractional release of intracellular Ca2+ (Lukyanenko et al. 1996; Sitsapesan & Williams, 1997; Xu & Meissner, 1998). These considerations suggest that the acute effects of T3 may be quite prominent under physiological conditions. The present results also show that the increase in peak INa was specific to the actions of T3. T4 and rT3, other hormones produced by the thyroid gland, produced no effect on INa. This type of specificity has been reported for the acute effects of T3 (but not rT3) to increase the inward rectifier K+ current (Ik1) (Gotzsche, 1994).
The present results indicate that acute exposure to T3 does not increase INa through alterations in the gating kinetics of INa activation, inactivation or recovery from inactivation. In ventricular myocytes, single channel recordings showed that acute T3 exposure did not increase single Na+ channel current amplitude but rather increased the bursting behaviour of the channel, i.e. caused an increase in open probability (Dudley & Baumgarten, 1993). Similarly, in ventricular myocytes, acute T3 exposure increases the open probability of Ik1 without affecting channel kinetics (Sakaguchi et al. 1996). Based on these considerations and the present data, it seems likely that T3 exerts an increase in macroscopic Na+ current by increasing the open probability of the channel.
What is the mechanism by which T3 rapidly increases INa? The answer to this question is not entirely clear. It is known that T3 is taken up in neonatal cardiomyocytes by an energy-dependent carrier-mediated mechanism that may be partly dependent on the Na+ gradient (Everts et al. 2001). In addition, there are T3 receptors in the cytoplasm that may function distinctly from their role in gene transcription in the nuclear compartment (Baumann et al. 2001), although their functional role is not known. However, Dudley & Baumgarten (1993) clearly showed that in rabbit ventricular myocytes, the acute effect of T3 to increase Na+ channel activity is not mediated via second messenger signalling, and required that T3 be in close proximity to the extracellular face of the channel. These findings suggest some sort of direct effect of T3 on the channel and are consistent with the relatively rapid time course over which T3 increases INa.
Several of the present results indicate that T3 elevates [Ca2+]i. T3 increased cell shortening, and could elicit delayed after-depolarizations and spontaneous activity between stimulated beats, both manifestations of elevated SR Ca2+ content and release. Moreover, depletion of SR Ca2+ content and inhibition of SR Ca2+ release by ryanodine eliminated the arrhythmogenic effects of T3. Also, direct measurements showed that T3 increased caffeine-induced SR Ca2+ release, baseline [Ca2+]i and [Ca2+]i transient amplitude, consistent with elevated SR Ca2+ content and release. These findings are in agreement with the effects of acute T3 exposure to increase tension-contractile activity in isolated muscle cells (Snow et al. 1992; Mager et al. 1992), isolated perfused hearts (Tielens et al. 1996), and the intact animal (Gotzsche, 1994). The fact that T3 did not alter L-type Ca2+ current and that its effects on INa were unaltered by Ni2+, an inhibitor of ICa,T, suggests that the effects of T3 to increase [Ca2+]i are not mediated through Ca2+ influx via voltage-dependent Ca2+ channels. Moreover, the prominent effects of T3 to increase cell shortening and [Ca2+]i are not consistent with the modest effects of Ca2+ influx via ICa,T (Sipido et al. 1998; Zhou & January, 1998). Alternatively, increases in Na+ influx via INa can alter intracellular Na+ gradients and thereby reverse Na+-Ca2+ exchange, resulting in significant Ca2+ influx (Sipido et al. 1995). This mode of Ca2+ influx is consistent with the increase in baseline intracellular Ca2+ concentration (Lipp et al. 2002) and can account for the increased intracellular Ca2+ content and release induced by T3.
The present results are consistent with clinical findings seen in hyperthyroid patients who traditionally present with classic symptoms of increased heart rate, stroke volume, cardiac output and cardiac contractility along with potential atrial irregularities including atrial fibrillation (Klein & Levey, 2000; Klein & Ojamaa, 2001). The present results, therefore, indicate that the non-genomic effects of T3 may contribute substantially to the effects of thyroid hormone traditionally attributed to chronic, genomic mechanisms. With regard to pacemaker activity, pacemakers within the sinoatrial (SA) node (Lipsius & Vassalle, 1978) as well as right atrial pacemakers outside of the SA node (Rubenstein & Lipsius, 1989) are partially dependent on activation of TTX-sensitive INa. Therefore, these pacemaker cell types are potential targets for T3 action. Moreover, our laboratory (Hüser et al. 2000a), as well as others (Rigg & Terrar, 1996; Bogdanov et al. 2001), has shown that intracellular Ca2+ release is a key component underlying atrial pacemaker and SA node activity. Specifically, voltage-dependent activation of ICa,T triggers local SR Ca2+ release, which in turn stimulates Na+-Ca2+ exchange current to depolarize the pacemaker potential toward threshold (Hüser et al. 2000a). In fact, preliminary recordings from cat atrial pacemaker cells indicate that acute T3 exposure stimulates pacemaker activity via a TTX-sensitive mechanism (Y. G. Wang & S.L. Lipsius, unpublished observations). Furthermore, in rat neonatal atrial myocytes, T3 stimulates pacemaker rate and Na+-Ca2+ exchange current (Sun et al. 2001). By increasing [Ca2+]i (via stimulation of INa), acute effects of T3 can be expected to enhance the normal Ca2+-mediated mechanisms underlying SA node and right atrial pacemaker activities, thereby contributing to the sinus (atrial) tachycardia induced by elevated serum levels of T3 in patients with long-standing hyperthyroidism.
Acknowledgments
We thank Christine E. Rechenmacher (deceased) and Anne Pezalla for their expert technical assistance with these experiments. This research was supported by a grant from Boots (Knoll) Pharmaceuticals (S.L.L.); NIH grants HL63753 (S.L.L.) and HL62231 (L.A.B.).
References
- Baumann CT, Maruvada P, Hager GL, Yen PM. Nuclear cytoplasmic shuttling by thyroid hormone receptors, multiple protein interactions are required for nuclear retention. J Biol Chem. 2001;276:11237–11245. doi: 10.1074/jbc.M011112200. [DOI] [PubMed] [Google Scholar]
- Binah O, Rubinstein I, Gilate E. Effects of thyroid hormone on the action potential and membrane currents of guinea pig ventricular myocytes. Pflugers Arch. 1987;409:214–216. doi: 10.1007/BF00584774. [DOI] [PubMed] [Google Scholar]
- Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- Brent G A. The molecular basis of thyroid hormone action. New Eng J Med. 1994;331:847–854. doi: 10.1056/NEJM199409293311306. [DOI] [PubMed] [Google Scholar]
- Chopra IJ, Sabatino L. Nature and sources of circulating thyroid hormone. In: Braverman LE, Utiger RD, editors. The Thyroid. New York: Lippincott, Williams and Wilkins; 2000. pp. 121–135. [Google Scholar]
- Davis PJ, Davis FB. Acute cellular actions of thyroid hormone and myocardial function. Ann Thorac Surg. 1993;56:S16–23. doi: 10.1016/0003-4975(93)90550-2. [DOI] [PubMed] [Google Scholar]
- Dudley S C, Baumgarten C M. Bursting of cardiac sodium channels after acute exposure to 3,5,3′-triiodo-l-thyronine. Circ Res. 1993;73:301–313. doi: 10.1161/01.res.73.2.301. [DOI] [PubMed] [Google Scholar]
- Everts ME, Verhoeven FA, Bezstarostik K, Moerings EP, Hennemann G, Visser TJ, Lamers JM. Uptake of thyroid hormones in neonatal rat cardiac myocytes. Endocrinology. 2001;142:11–12. doi: 10.1210/endo.137.10.8828482. [DOI] [PubMed] [Google Scholar]
- Freedberg AS, Papp GJ, Vaughan Williams EM. The effect of altered thyroid state on atrial intracellular potentials. J Physiol. 1970;7:50–54. doi: 10.1113/jphysiol.1970.sp009066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzsche LBH. l-Triiodothyronine acutely increases Ca2+ uptake in the isolated perfused rat heart. Changes in L-type Ca2+ channels and β-receptors during short and long term hyper- and hypothyroidism. Europ J Endo. 1994;130:171–179. doi: 10.1530/eje.0.1300171. [DOI] [PubMed] [Google Scholar]
- Harris DR, Green WL, Craelius W. Acute thyroid hormone promotes slow inactivation of sodium current in neonatal cardiac myocytes. Biochem Biophys Acta. 1991;1045:175–181. doi: 10.1016/0167-4889(91)90081-8. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüser J, Blatter LA, Lipsius SL. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. 2000a;524:415–422. doi: 10.1111/j.1469-7793.2000.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüser J, Wang YG, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol. 2000b;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss E, Jakob G, Kranias EG, Edes I. Thyroid hormone-induced alteration in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ Res. 1995;75:245–251. doi: 10.1161/01.res.75.2.245. [DOI] [PubMed] [Google Scholar]
- Klein I, Levey GS. The cardiovascular system in thyrotoxicosis. In: Braverman LE, Utiger RD, editors. The Thyroid. New York: Lippincott, Williams and Wilkins; 2000. pp. 596–604. [Google Scholar]
- Klein I, Ojamaa K. Mechanisms of Disease: Thyroid hormone and the cardiovascular system. New Eng J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- Lipp P, Egger M, Niggli E. Spatial characteristics of sarcoplasmic reticulum Ca2+ release events triggered by L-type Ca2+ current and Na+ current in guinea-pig cardiac myocytes. J Physiol. 2002;542:383–393. doi: 10.1113/jphysiol.2001.013382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp P, Niggli E. Sodium current-induced calcium signals in isolated guinea-pig ventricular myocytes. J Physiol. 1994;474:439–446. doi: 10.1113/jphysiol.1994.sp020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsius SL, Vassalle M. Dual excitatory channels in the sinus node. J Mol Cell Cardiol. 1978;10:753–767. doi: 10.1016/0022-2828(78)90409-1. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Gyorke I, Gyorke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflugers Arch. 1996;432:1047–1054. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- Mager S, Palti Y, Binah O. Mechanism of hyperthyroidism-induced modulation of the L-type Ca2+ current in guinea pig ventricular myocytes. Pflugers Arch. 1992;421:425–430. doi: 10.1007/BF00370252. [DOI] [PubMed] [Google Scholar]
- Rigg L, Terrar DA. Possible role of calcium release from sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- Rubinstein I, Binah O. Thyroid hormone modulates membrane currents in guinea-pig ventricular myocytes. Archiv Pharmacol. 1989;340:705–711. doi: 10.1007/BF00717748. [DOI] [PubMed] [Google Scholar]
- Rubinstein DS, Lipsius SL. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res. 1989;64:648–657. doi: 10.1161/01.res.64.4.648. [DOI] [PubMed] [Google Scholar]
- Sakaguchi Y, Cui G, Sen L. Acute effects of thyroid hormone on inward rectifier potassium channel currents in guinea pig ventricular myocytes. Endocrinology. 1996;137:4744–4751. doi: 10.1210/endo.137.11.8895342. [DOI] [PubMed] [Google Scholar]
- Segal J. Action of the thyroid hormone at the level of the plasma membrane. Endocrinol Res. 1989;15:619–649. doi: 10.3109/07435808909036355. [DOI] [PubMed] [Google Scholar]
- Segal J. Calcium is the first messenger for the action of thyroid hormone at the level of the plasma membrane: first evidence for an acute effect of thyroid hormone on calcium uptake in the heart. Endocrinology. 1990;126:2693–2702. doi: 10.1210/endo-126-5-2693. [DOI] [PubMed] [Google Scholar]
- Shenoy R, Klein I, Ojamaa K. Differential regulation of SR calcium transporters by thyroid hormone in rat atria and ventricles. Am J Physiol Heart Circ Physiol. 2001;281:H1690–1696. doi: 10.1152/ajpheart.2001.281.4.H1690. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Severson DL. Thyroid status and potassium current in rat ventricular myocytes. Am J Physiol. 1995;268:H576–583. doi: 10.1152/ajpheart.1995.268.2.H576. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Carmeliet E, Pappano A. Na+ current and Ca2+ release from the sarcoplasmic reticulum during action potentials in guinea-pig ventricular myocytes. J Physiol. 1995;489:1–17. doi: 10.1113/jphysiol.1995.sp021025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Carmeliet E, Van De Werf F. T-type Ca2+ current as a trigger for Ca2+ release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. J Physiol. 1998;508:439–451. doi: 10.1111/j.1469-7793.1998.439bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of current flow through ryanodine receptors by luminal Ca2+ J Mem Biol. 1997;159:179–185. doi: 10.1007/s002329900281. [DOI] [PubMed] [Google Scholar]
- Snow TR, Deal MT, Connelly TS, Yokoyama Y, Novitzky D. Acute inotropic responses of rabbit papillary muscle to triiodothyronine. Cardiology. 1992;80:112–117. doi: 10.1159/000174988. [DOI] [PubMed] [Google Scholar]
- Sun Z, Ojamaa K, Coetzee WA, Artman M, Klein I. Effects of thyroid hormone on action potential and repolarizing currents in rat ventricular myocytes. Am J Physiol Endocrinol Metab. 2000;278:E302–307. doi: 10.1152/ajpendo.2000.278.2.E302. [DOI] [PubMed] [Google Scholar]
- Sun ZQ, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J Mol Cell Cardiol. 2001;33:811–824. doi: 10.1006/jmcc.2001.1353. [DOI] [PubMed] [Google Scholar]
- Tielens ET, Forder JR, Chatham JC, Marrelli SP, Ladenson PW. Acute l-triiodothyronine administration potentiates inotropic responses to β-adrenergic stimulation in the isolated perfused rat heart. Cardiovasc Res. 1996;32:306–310. doi: 10.1016/0008-6363(96)00096-x. [DOI] [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine activates a glibenclamide-sensitive K+ current in cat atrial myocytes. Am J Physiol. 1995;268:H1322–1334. doi: 10.1152/ajpheart.1995.268.3.H1322. [DOI] [PubMed] [Google Scholar]
- Xu L, Meissner G. Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum luminal Ca2+ Biophys J. 1998;75:2302–2312. doi: 10.1016/S0006-3495(98)77674-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, January CT. Both T- and L-type Ca2+ channels can contribute to excitation-contraction coupling in cardiac Purkinje cells. Biophys J. 1998;74:1830–1839. doi: 10.1016/S0006-3495(98)77893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Lipsius SL. T-type calcium current in latent pacemaker cells isolated from cat right atrium. J Mol Cell Cardiol. 1994;26:1211–1219. doi: 10.1006/jmcc.1994.1139. [DOI] [PubMed] [Google Scholar]