

Abstract
Intracellular calcium ([Ca2+]i) and isometric force were measured in left ventricular (LV) trabeculae from spontaneously hypertensive rats (SHR) with failing hearts and normotensive Wistar-Kyoto (WKY) controls. At a physiological stimulation frequency (5 Hz), and at 37 °C, the peak stress of SHR trabeculae was significantly (P ≤; 0.05) reduced compared to WKY (8 ± 1 mN mm−2(n = 8)vs. 21 ± 5 mN mm−2(n = 8), respectively). No differences between strains in either the time-to-peak stress, or the time from peak to 50 % relaxation were detected. Measurements using fura-2 showed that in the SHR both the peak of the Ca2+ transient and the resting [Ca2+]i were increased compared to WKY (peak: 0.69 ± 0.08 vs. 0.51 ± 0.08 μm (P ≤ 0.1) and resting: 0.19 ± 0.02 vs. 0.09 ± 0.02 μm (P ≤ 0.05), SHR vs. WKY, respectively). The decay of the Ca2+ transient was prolonged in SHR, with time constants of: 0.063 ± 0.002 vs. 0.052 ± 0.003 s (SHR vs. WKY, respectively). Similar results were obtained at 1 Hz stimulation, and for[Ca2+]o between 0.5 and 5 mm. The decay of the caffeine-evoked Ca2+ transient was slower in SHR (9.8 ± 0.7 s (n = 8)vs. 7.7 ± 0.2 s (n = 8) in WKY), but this difference was removed by use of the SL Ca2+-ATPase inhibitor carboxyeosin. Histological examination of transverse sections showed that the fractional content of perimysial collagen was increased in SHR compared to WKY (18.0 ± 4.6 % (n = 10)vs. 2.9 ± 0.9 % (n = 11) SHR vs. WKY, respectively). Our results show that differences in the amplitude and the time course of the Ca2+ transient between SHR and WKY do not explain the reduced contractile performance of SHR myocardium per se. Rather, we suggest that, in this animal model of heart failure, contractile function is compromised by increased collagen, and its three-dimensional organisation, and not by reduced availability of intracellular Ca2+.
Alterations to cellular Ca2+ homeostatic mechanisms are often implicated in the pathophysiology of heart disease. A number of studies have reported abnormal myocyte Ca2+ handling during the heart failure that follows a prolonged period of pressure overload. In studies using tissue from either human (Gwathmey et al. 1987; Beuckelmann et al. 1992; Mattiello et al. 1998; Dipla et al. 1999) or animal (Bing et al. 1991; Brooks et al. 1994; Gómez et al. 1997) models, the impaired mechanical performance of hearts in failure has been associated with various defects. These have included remodelling of the ventricular chamber, extracellular matrix hyperplasia, loss of myocytes by apoptosis, and decreased myocyte contractile function. Experimental models have employed a range of different species as well as different myocardial preparations. Results from these studies have generally found that, along with the contractile dysfunction, there are corresponding alterations in the Ca2+ transport mechanisms associated with excitation-contraction coupling (for a review see Movsesian & Schwinger, 1998).
The spontaneously hypertensive rat (SHR), introduced by Okamoto & Aoki in 1963, is an animal model of genetic systemic hypertension in which the development of hypertension leads to heart failure. The progression of disease in SHR follows a predictable pattern, with hypertension being the primary stimulus for the development of hypertrophy. End-stage heart failure routinely develops in SHR between 18 and 24 months of age (Perreault et al. 1990; Bing et al. 1991; Brooks et al. 1994), following a long period of stable hypertrophy, during which contractile function is preserved or enhanced (Conrad et al. 1991). Furthermore, unlike most other animal models, the myocardial changes due to senescence that usually accompany the human disease are mimicked in SHR (Assayag et al. 1997; Fitzsimons et al. 1999). We therefore chose the SHR as our model of heart failure, using Wistar-Kyoto rats of a similar age as normotensive controls.
A major problem in studying isolated cardiac muscle is the need to minimise metabolic demand in the preparation, which is not generally perfused. For this reason, experiments are commonly carried out at low temperatures and low stimulation frequencies. It is therefore possible that cellular processes observed under these conditions may not be representative of those in vivo. The need to reduce stimulation frequency and temperature in order to avoid metabolic insufficiency can be obviated by the use of cardiac trabeculae. These preparations are structurally homologous to ventricular wall tissue, being made up predominantly of longitudinally arranged myocytes surrounded by a narrow rim of endothelial cells (Hanley et al. 1999). Although isolated myocytes provide minimal diffusion distances, they were not used in this study, since they do not permit quantitative force measurements at working sarcomere lengths. The axial alignment of myocytes in trabeculae make them ideal preparations for simultaneous measurement of isometric force and [Ca2+]i. Using this preparation we have found that although force is reduced, resting [Ca2+]i and the amplitude of the Ca2+ transient are increased, showing a dissociation between contractile function and [Ca2+]i availability.
Methods
Experimental animals and haemodynamic measurements
Spontaneously hypertensive rats (SHR) and normotensive Wistar-Kyoto rats (WKY) of both sexes were obtained at 6 weeks of age and were housed under control conditions with ad libitum food and water. The SHR progresses to heart failure with senescence, and after ≈20 months of age signs of worsening myocardial performance (such as respiratory distress, weight loss, lethargy, poor grooming) could be detected. As soon as overt signs of heart failure were apparent, SHR were used for experimentation. Systolic blood pressure and heart rate were measured with a tail cuff sphygmomanometer (Model 179, IITC Inc., Life Science Instruments, Woodland Hills, CA, USA), and animals were weighed, anaesthetised with halothane and killed by decapitation. Experimental procedures were approved by the Animal Ethics Committee of the University of Auckland. Healthy WKY control animals of comparable age were selected, and subjected to the same experimental protocols. The diagnosis of heart failure in SHR was confirmed by post mortem examination. In some cases, M-mode echocardiography was performed on lightly anaesthetised (20 mg kg−1i.p. of Teletamine HCl and Zolazepam HCl; Vibrac Laboratories (NZ) Ltd) SHR to confirm the signs of failure, and on matched WKY controls. Analysis of the M-mode data confirmed ventricular dilatation (end diastolic volume, SHR: 0.369 ± 0.036 μl (n = 3), and WKY: 0.174 ± 0.043 μl (n = 4), P ≤ 0.05) and reduced fractional shortening in SHR compared to WKY (SHR: 0.34 ± 0.05 (n = 3), WKY: 0.56 ± 0.04 (n = 4), P ≤ 0.05).
Dissection and mounting of left ventricular trabeculae
Perfusion of the coronary circulation with oxygenated dissection solution (see below) was maintained throughout dissection. When present, an unbranched, cylindrical trabecula (average length 1.9 ± 0.1 mm, cross-sectional area 0.038 ± 0.003 mm2, n = 56) from the left ventricle was dissected free with a small block of ventricular tissue at either end. The trabecula was then transferred to a Perspex bath (volume 450 μl) on the stage of an inverted microscope (Nikon Diaphot 300, Japan), as described previously (Hanley & Loiselle, 1998). One end of the trabecula was mounted in a wire cradle extending from the silicon beam of a force transducer (model AE801, SensoNor, Horten, Norway) while the other end of the trabecula was held in a monofilament (30 μm diameter) nylon snare protruding from a stainless steel tube attached to a micromanipulator. Trabeculae were continuously superfused with oxygenated modified Krebs-Henseleit solution (see below) at a flow rate of 7 ml min−1. Field stimulation (5 ms pulse) was applied at 0.1 Hz by a Digitimer D100 (Digitimer, Welwyn Garden City, Herts, UK), using platinum electrodes. The central portion of the trabecula was viewed using a × 40 objective (NA 0.55) and a charge-coupled device camera connected to a video monitor to allow striations to be observed. Trabeculae were adjusted to a sarcomere length of 2.1-2.2 μm. This sarcomere length has previously been shown to be comparable to that maximally attained in vivo (Rodriguez et al. 1992) such as might occur at the end of diastolic filling.
Fura-2 AM (100 μg; Texas Fluorescent Laboratories, Austin, TX, USA) was dissolved in 30 μl of freshly prepared anhydrous dimethyl sulphoxide (DMSO; Aldrich) with 5 % w/v pluronic F127, and added to 10 ml of Krebs-Henseleit superfusate ([Ca2+]o 1 mm). Oxygenated loading solution was continuously circulated for a period of 2 h, at room temperature (20-22 °C), whilst stimulating the trabeculae at a frequency of 0.1 Hz. After loading was complete, the superfusate was switched to Krebs-Henseleit solution to remove any extracellular remnants of the loading solution.
Chemicals and solutions
Trabeculae were superfused with a modified Krebs-Henseleit solution containing (mm): 118 NaCl, 4.75 KCl, 1.18 MgSO4, 1.18 KH2PO4, 24.8 NaHCO3 and 10 d-glucose. The solutions were continuously bubbled with 95 % O2 and 5 % CO2; pH was maintained at 7.4.[Ca2+]o was adjusted, as required, by addition of CaCl2 from a 1 m stock solution. The dissection solution contained 0.25 mm Ca2+ plus 20 mm 2,3-butanedione monoxime (BDM) to protect the myocardium from a high rate of energy utilisation, by reducing cross-bridge cycling (Mulieri et al. 1989) and SR content (Steele & Smith, 1993; Adams et al. 1998). On removal of BDM from the superfusate,[Ca2+]o was increased gradually to 1 mm. To maximise retention of indicator in the cytoplasm at 37 °C, 1 mm probenecid was added to all solutions following fura-2 loading (Di Virgilio et al. 1990). A modified Tyrode solution was used for a number of trabeculae in which caffeine contractures were carried out in both normal Na+ (143 mm) and Na+-free solutions. We used 5,6-carboxyeosin diacetate (CE, Molecular Probes) to inhibit the sarcolemmal (SL) Ca2+-ATPase (Choi & Eisner, 1999). Trabeculae were loaded for 30 min with 20 μm CE followed by > 10 min period for de-esterification. The Tyrode solution was composed of the following (mm): 141.8 NaCl, 6 KCl, 1.2 MgSO4.7H20, 1.2 Na2HPO4, 10 Hepes, 2 CaCl2 and 10 d-glucose, adjusted to pH 7.4 with NaOH. A Na+-free and Ca2+-free Tyrode solution was obtained by equimolar substitution of NaCl with LiCl, and the use of K2HPO4.3H2O in the place of Na2HPO4. CaCl2 was replaced with 1 mm EGTA, and the pH was adjusted with KOH. Tyrode solutions were continuously bubbled with 100 % oxygen. All chemicals used were purchased from Sigma (Sigma Aldrich, Australia) unless otherwise stated.
Measurement of [Ca2+]i using fura-2
Trabeculae were illuminated using a 75 W xenon arc lamp, and a spectrophotometric system (Cairn Research, Faversham, Kent, UK) that provided rapidly alternating excitation wavelengths (340, 360 and 380 nm) for ratiometric measurement of fura-2 fluorescence at 500 nm, as previously described (Hanley & Loiselle, 1998). An adjustable window restricted the area of illumination to a rectangle of approximately 100 μm × 250 μm over the central portion of the trabecula.
Subtraction of tissue autofluorescence, measured at room temperature prior to loading, was made for the 340 and 380 nm excitation wavelengths before obtaining the ratio. No evidence of temperature-dependent changes in autofluorescence was detected in a representative sample of trabeculae (data not shown). A component of autofluorescence originates from the reduced nicotinamide adenine nucleotides (NADH > NADPH) and oxidised flavoproteins (Aubin, 1979; Brandes & Bers, 1996) since their emission spectra partly overlap the fura-2 emission spectrum. However, we found no difference in autofluorescence between SHR and WKY preparations (340 nm/380 nm ratio, SHR: 0.73 ± 0.03 (n = 24), WKY: 0.77 ± 0.02 (n = 32)). It therefore follows that changes in fura-2 fluorescence ratios could not be attributed to some difference in autofluorescence between SHR and WKY trabeculae. After loading with fura-2, the mean fluorescence at 360 nm excitation wavelength was increased to 5.4 ± 0.3 times the autofluorescence value. Subsequent membrane permeabilisation at the end of experimentation showed that compartmentalised fura-2 accounted for less than 15 % of the total fluorescence (data not shown).
Calibration of fura-2
The Ca2+ concentration corresponding to a given fura-2 fluorescence ratio was estimated by applying the following equation (Grynkiewicz et al. 1985):
| (1a) |
where Rmax and Rmin are the 340 nm/380 nm fluorescence ratios for saturating and zero Ca2+ solutions, respectively; β is the ratio of the emitted fluorescence at 380 nm excitation in the Ca2+-free solution over that at 380 nm in the saturating solution; and R is the observed 340 nm/380 nm fluorescence ratio. No correction for fura-2 kinetics was applied. A full in vivo calibration procedure was not undertaken, but measurements of Rmin and Rmax were made at 37 °C for a number of trabeculae. A value for Kd (a simple scaling factor in eqn (1a)) of 371 nm for fura-2 in cardiac myocytes at 37 °C was obtained from the literature (Haworth & Redon, 1998).
We noted that the rate of loss of intracellular fura-2 increased as the temperature was increased above room temperature, even in the presence of an anionic transport blocker (probenecid 1 mm). At 37 °C ≈20 % of the indicator was lost per hour (n = 6). While this effect is of little consequence when making ratiometric measurements, when in vivo calibrations were performed the 380 nm signal used to obtain β in eqn (1a) would be affected by the loss of indicator since equilibration to the minimum and maximum [Ca2+] took ≈30 min in these multicellular preparations. We therefore used the method proposed by Jiang & Julian (1997) to account for changes in the 380 nm signal with time due to the loss of indicator during measurement of Rmax. We multiplied the 380 nm fluorescence at Rmax by the ratio of the fluorescence at the isosbestic wavelength (360 nm) for fura-2 at the beginning (Rmin) and at the end (Rmax) of the calibration procedure:
| (1b) |
Values for Rmin, Rmax and β were obtained by exposing RV trabeculae from Wistar rats to a Ca2+-free (Rmin) solution followed by a saturating Ca2+ (Rmax) solution. The calibration solution for Rmin contained (mm): 118 NaCl, 6 KCl, 1.18 MgSO4, 1.18 KH2PO4, 24.8 NaHCO3, 10 glucose, 10 caffeine, 10 BDM, 10 EGTA, 0.1 2,5-di(tert-butyl)-1,4-benzohydroquinone (TBQ) and 1 ouabain. The corresponding solution for Rmax was: 120 LiCl, 1.18 MgSO4, 1.18 KH2PO4, 20 NaHCO3, 4.82 KHCO3, 10 glucose, 10 caffeine, 10 BDM, 0.1 TBQ, 1 ouabain and 0.01 Br-A23187. These procedures provided the following values for use with eqn (1a): β = 2.7, Rmin = 0.25 and Rmax = 2.5.
Fura-2 fluorescence is sensitive, in part, to the intracellular environment. Since this might be different between the rat strains used in this study, emitted fluorescence for the 380 nm vs. 340 nm excitation wavelengths was plotted separately for a number of SHR and WKY trabeculae and for in vitro calibration data (Bakker et al. 1993). The slopes of these relationships were determined by linear regression: comparison of these slopes revealed no significant difference between SHR (-0.39 ± 0.06, n = 13), WKY (-0.35 ± 0.04, n = 9) and in vitro calibration data (-0.37 ± 0.04, n = 8). We therefore concluded that the Rmin and Rmax values obtained from our in vivo calibrations could be reliably applied to both rat strains, and that there was no strain-dependent influence on the recorded fluorescence ratios.
Dissection of sarcolemmal transport mechanisms
Investigation of the Ca2+ fluxes contributing to the decay of [Ca2+]i was made for a number of trabeculae by functionally eliminating the SR Ca2+ transport with 10 mm caffeine, and by subsequent exposure to CE, and 0 mm [Na+]o combined with 10 mm Ni+, to inhibit the SL Ca2+-ATPase and Na+-Ca2+ exchange, respectively (Choi & Eisner, 1999). To eliminate any SL Ca2+ influx during the evaluation of SL Ca2+ extrusion, a 0 mm Ca2+ Tyrode solution was used as the superfusate (O'Neill & Eisner, 1990). These experiments were carried out at room temperature (20-22 °C): (1) to slow the time course of Ca2+ transport compared to caffeine diffusion across the multicellular trabeculae, (2) to minimise any possible difference in SR Ca2+ buffering during futile cycling in the presence of caffeine, and (3) to increase the relative contribution of the Ca2+-ATPase compared to Na+-Ca2+ exchange.
Fixation of trabeculae and staining for collagen
A number of preparations were fixed for morphological examination on completion of experiments. Trabeculae were first stretched by 5 % (to allow for shrinkage during the fixation process) and fixed with Bouin's solution, as previously described (Hanley et al. 1999). Trabeculae were cut in half, and one half was exposed to picrosirius red (PSR) (0.1 % w/v sirius red F3BA in saturated picric acid) for 2 h. Both halves were then resin-embedded, and the second half was stained with methyl blue (1 %) for myocytes and basic fuchsin (1 %) for the extracellular matrix (see Fig. 5 legend). The PSR stained preparations were imaged using a confocal microscope (Leica TCS 4D, Heidelberg, Germany) with a × 25 objective (0.75 NA). By summing a series of confocal sections, extended-focus images to a depth of 50 μm were obtained for qualitative analysis of the longitudinal collagen distribution. Transmitted light images of the transversely sectioned trabeculae were captured with a digital camera (Zeiss ProgRes 3008) mounted on a light microscope (Leica DM R, Wetzla, Germany) with a × 100 objective (1.4 NA). Images were then analysed to determine the relative proportions of myocytes and collagen using NIH Image (http://rsb.info.nih.gov/nih-image/). Total cross-sectional area calculated in this way excluded unstained areas (such as the interior of blood vessels).
Figure 5. Morphometric analysis of LV trabeculae.
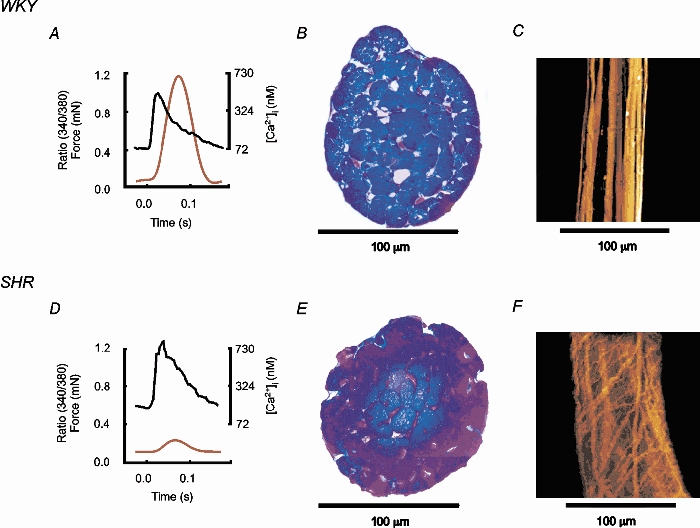
Examples of force, [Ca2+]i (5 Hz frequency, 2 mm[Ca2+]o, 37 °C, sarcomere length: 2.1 μm) and collagen distribution in representative LV trabeculae of similar dimensions from WKY (top) and SHR (bottom). A and D, averaged 340 nm/380 nm ratios and force. B and E, transversely sectioned trabeculae, stained blue for myocytes and pink for collagen. WKY trabecula: 2 % collagen, SHR trabecula: 7 % collagen. C and F, trabeculae in longitudinal section (extended focus to a depth of 50 μm) stained for collagen.
Data analysis
Unbiased measurement of Ca2+ transient and isometric force parameters were obtained from data averaged over a number of cardiac cycles, using a custom-written analysis program (IDL, Research Systems Inc., Boulder, CO, USA) (see Fig. 1). A five-parameter exponential function was used to describe the Ca2+ transient:
| (2) |
Figure 1. Example of data analysis.
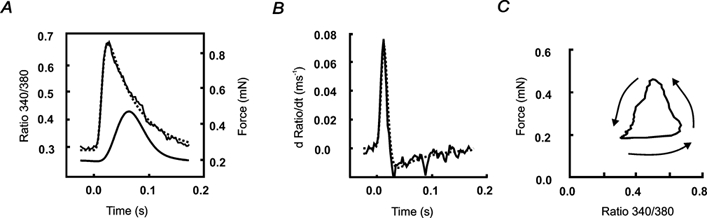
Averaged data from 19 consecutive cardiac cycles from a WKY LV trabecula, at a stimulation frequency of 5 Hz (2 mm[Ca2+]o, 37 °C). A, averaged fura-2 fluorescence and force transients, with exponential function fitted (dotted line) to the fluorescence transient according to eqn (2). The stimulus was applied at time 0. The time constant of decay in fluorescence for this trabecula, obtained from the fitted curve, was 0.054 s, and the time from peak force to 50 % relaxation was 0.033 s. B, the time-derivative of fluorescence from A for both the averaged data and the fitted function (dotted line). The maximum rate of rise of fluorescence was obtained from the peak of the time-derivative. C, force as a function of the 340 nm/380 nm fluorescence ratio for the data shown in A. The arrows indicate the direction of the phase plot with time.
Typical values of parameters found using eqn (2) were: A0 1.3 (maximum fluorescence 340 nm/380 nm ratio change); A1 0.1 s (time constant of exponential decline during relaxation); A2 1 ms (offset in time between rising and decaying phases of the Ca2+ transient); A3 3 ms (the time constant of rise of the transient); A4 0.4 (minimum fluorescence before stimulation). Peak systolic fluorescence, minimum resting fluorescence, time-to-peak fluorescence, and the maximum rate of rise of fluorescence (from the differential), were obtained from both the averaged and fitted data (see Fig. 1). Note that resting (or diastolic) Ca2+ was calculated from the minimum fluorescence in the interval between transients. Force was also averaged for the same number of cardiac cycles, and measurements made of the peak twitch force, the resting (diastolic) force, the time-to-peak force, the maximum rate of rise of force and the time-to-50 % relaxation.
Results from the averaged data were imported directly from the IDL analysis program into statistical analysis software (SAS Institute, Cary, NC, USA) where analysis of variance (ANOVA) was carried out using its general linear model procedure. Data are expressed as means ± s.e.m., and statistical significance as P ≤ 0.05. In total, 27 SHR and 33 WKY LV trabeculae were used in this study.
Results
Changes associated with heart failure
Physical evidence for hypertrophy and heart failure in the SHR is summarised in Table 1, together with corresponding data for the WKY control animals. Measured values of systolic blood pressure confirmed that the SHR were hypertensive (191 ± 6 mmHg), and that the control WKY animals were normotensive (129 ± 3 mmHg). Measurements showed that SHR animals had developed cardiac hypertrophy which included the RV, increased ratio of lung wet weight to dry weight (pulmonary oedema), and increased lung wet weight to body weight ratio. Other indicators of heart failure, such as localised pulmonary haemorrhages, the presence of left atrial thrombi and visible myocardial fibrosis, were all frequently noted in SHR. These observations are consistent with previous reports of SHR in heart failure (Conrad et al. 1991; Bing et al. 1995). As noted in Methods, measurements of reduced shortening by echocardiography also support the idea that the SHR hearts were failing.
Table 1.
Changes associated with heart failure in SHR
| Parameter | WKY Male | WKY Female | SHR Male | SHR Female | P ≤ 0.05 |
|---|---|---|---|---|---|
| Number of animals | 21 | 12 | 17 | 10 | |
| Age (months) | 24.2 ± 0.5 | 24.1 ± 0.4 | 21.0 ± 0.4 | 23.2 ± 0.3 | |
| Body wt (g) | 424 ± 8 | 281 ± 10 | 381 ± 15 | 237 ± 7 | †* |
| Systolic blood pressure (mmHg) | 131 ± 4 | 127 ± 4 | 189 ± 5 | 197 ± 14 | † |
| Heart rate (min−1) | 347 ± 11 | 374 ± 7 | 406 ± 10 | 445 ± 7 | †* |
| Tibial length (cm) | 5.3 ± 0.1 | 4.8 ± 0.0 | 5.5 ± 0.1 | 5.0 ± 0.1 | * |
| Outer diameter of aorta (mm) | 2.7 ± 0.1 | 2.6 ± 0.1 | 3.6 ± 0.1 | 3.4 ± 0.1 | † |
| LV wt/body wt (mgg−1) | 1.73 ± 0.06 | 1.76 ± 0.12 | 2.50 ± 0.18 | 3.29 ± 0.19 | †* |
| RV wt/body wt (mgg−1) | 0.57 ± 0.02 | 0.61 ± 0.03 | 0.65 ± 0.06 | 0.75 ± 0.04 | † |
| Septal wt/body wt (mgg−1) | 1.06 ± 0.04 | 1.42 ± 0.12 | 2.04 ± 0.12 | 2.20 ± 0.24 | †* |
| Lung wt/body wt (mgg−1) | 4.03 ± 0.34 | 4.58 ± 0.15 | 6.67 ± 0.56 | 6.90 ± 0.43 | † |
| Lung wet/dry wt | 4.96 ± 0.03 | 4.87 ± 0.06 | 5.59 ± 0.18 | 5.60 ± 0.16 | † |
| Liver wt/body wt | 35.19 ± 1.68 | 33.81 ± 1.10 | 43.31 ± 1.49 | 44.43 ± 1.57 | † |
Values are means ± s.e.m. WKY: normotensive Wistar-Kyoto rats; SHR: spontaneously hypertensive rats with heart failure; LV: left ventricular; RV: right ventricular; wt: wet weight. Symbols denote P≤ 0.05:
difference between sex
difference between rat strains.
Both male and female animals were used in this study, and analysis of data showed the expected sex differences relating to body size. For both strains of rat, heart rate was higher in females than in males. Heart rate was also increased in SHR when compared to WKY, but the sex differences in heart rate were the same for both rat strains. Body weight was lower in SHR (for both male and female), although tibial length (an index of body size) did not differ between SHR and WKY. Sex-related differences were not found for any of the variables associated with either the mechanical or the fluorescence measurements. Data from both male and female animals were therefore combined for experiments involving subgroups of the total population.
Force and [Ca2+]i at a physiological stimulation frequency
A stimulation frequency of 5 Hz was chosen to mimic the normal resting heart rate in rats. Figure 1 shows an example of averaged data obtained from a WKY LV trabecula stimulated at 5 Hz. The fitted fluorescence transient function (see eqn (2)) is superimposed on the averaged fluorescence transient for these data (Fig. 1A dotted line), as is the fitted differential (Fig. 1B dotted line). From these data it is apparent that the equation used to describe the fura-2 fluorescence transient is sufficient to capture the time course of the Ca2+ transient, and provides a bias-free measure of the major features of the Ca2+ transient. Note (Fig. 1A) that for WKY at 37 °C, even at 5 Hz stimulation frequency, there is a return to resting levels of both force and Ca2+, indicated in the rest interval preceding time 0 for the averaged data. Data from LV trabeculae at a stimulation frequency of 5 Hz are summarised in Table 2. It is apparent that the minimum resting and peak systolic fluorescence were increased in SHR compared to WKY, with no difference in the time-to-peak fluorescence between strains. Despite the increased amplitude and time course of the SHR Ca2+ transient, twitch force was reduced. Peak force normalised to cross-sectional area (stress) was only 40 % of that in WKY. The kinetics of the force response appeared similar between strains, since there was no difference in the time-to-peak stress or in the time-to-50 % relaxation. The reduced twitch amplitude, and lack of difference in the time-to-peak stress are in good agreement with the results of Bing et al. (1991) although these authors did not report any changes in the amplitude of the Ca2+ transient. In addition, Bing et al. (1991) reported that the resting [Ca2+]i was lower in SHR in comparison to age-matched WKY. In view of the lower stimulation frequency and[Ca2+]o used by Bing et al. (1991) we performed further experiments to examine whether these differences could be explained by the different experimental conditions.
Table 2.
Comparison of [Ca2+]i, and force between LV trabeculae from failing SHR hearts am normotensive WKY
| Variable | WKY | SHR | P ≤ 0.05 |
|---|---|---|---|
| No. of animals | 8 | 8 | |
| Trabeculae cross-sectional area (mm2) | 0.050 ± 0.004 | 0.059 ± 0.008 | |
| Time-to-peak 340/380 ratio (s) | 0.035 ± 0.003 | 0.040 ± 0.002 | |
| Time constant of 340/380 ratio decay (s) | 0.052 ± 0.003 | 0.063 ± 0.002 | * |
| Resting 340/380 ratio | 0.43 ± 0.03 | 0.60 ± 0.03 | * |
| Resting [Ca2+]i, (μm) | 0.09 ± 0.02 | 0.19 ± 0.02 | * |
| Peak 340/380 ratio | 0.98 ± 0.08 | 1.16 ± 0.06 | P = 0.1 |
| Peak [Ca2+]i, (μm) | 0.51 ± 0.08 | 0.69 ± 0.08 | P = 0.1 |
| Maximum rate-of-rise 340/380 ratio (ms−1) | 0.08 ± 0.01 | 0.08 ± 0.01 | |
| Time-to-peak force (s) | 0.067 ± 0.002 | 0.068 ± 0.004 | |
| Time-to-50% relaxation of force (s) | 0.031 ± 0.001 | 0.032 ± 0.001 | |
| Resting stress (mN mm−2) | 3.9 ± 1.4 | 3.6 ± 0.8 | |
| Peak systolic stress (mN mm−2) | 20.9 ± 4.8 | 8.2 ± 1.1 | * |
| Active stress (mN mm−2) | 16.9 ± 4.6 | 4.6 ± 1.4 | * |
| Peak 340/380 ratio to peak force (s) | 0.032 ± 0.004 | 0.028 ± 0.004 |
Data are means ± s.e.m. from LV trabeculae at 37°C, stimulated at a frequency of 5 Hz, and with [Ca2+]0 of 2 mm. The 340 nm/380 nm fura-2 fluorescence ratio (340/380 ratio) and force were averaged for a number of consecutive cardiac cycles (∼16), and the Ca2+ transient fitted according to eqn (2) (see text). Variables in the table associated [Ca2+]i were derived from the fitted equation, and were no different from those measured from the averaged transient (not shown). Statistical analysis revealed no sex differences for any of the above variables. Means are therefore from both male and female animals.
P < 0.05.
Effect of extracellular [Ca2+]
It is well known that changes in extracellular Ca2+ ([Ca2+]o) have profound effects on force production and [Ca2+]i homeostasis (Allen et al. 1976). Increased[Ca2+]o results in an increase in the amount of Ca2+ released from the SR (Bers, 1989), which can explain the increase in the magnitude of the Ca2+ transient. We therefore examined the effect of altering[Ca2+]o over a range that encompasses that used in most other experiments reported in the literature. To minimise the risk of Ca2+ overload,[Ca2+]o was increased only until the magnitude of the twitch started to decrease. In addition, a stimulation frequency of 1 Hz was used to mimic the low rates that have generally been used in other investigations.
The effects of altering[Ca2+]o are summarised in Fig. 2 and Fig. 3. Sample fluorescence and force records at 1.5 mm[Ca2+]o are shown in Fig. 2A for WKY (left) and SHR (right). Note that at 1 Hz stimulation frequency, there is a complete return to resting levels of both [Ca2+]i and force between stimuli for both rat strains.
Figure 2. The effect of[Ca2+]o on [Ca2+]i and stress.
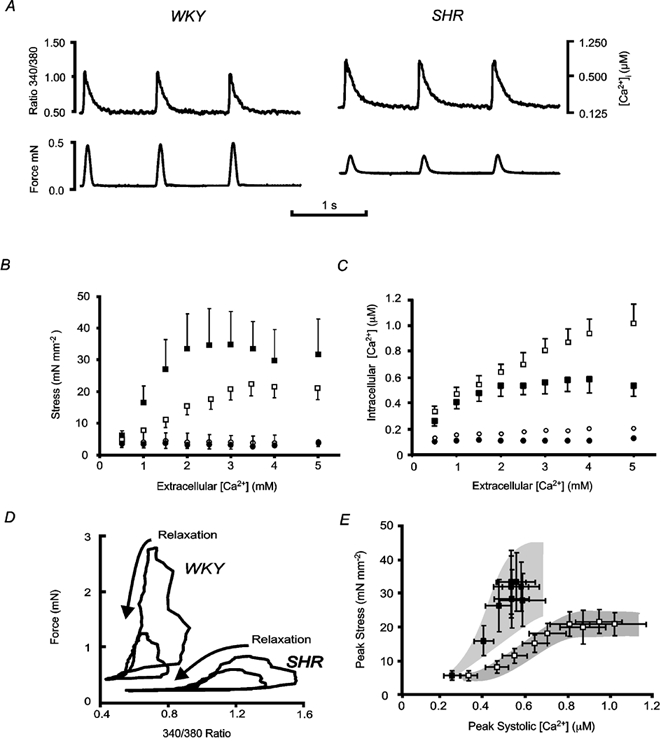
Representative (A and D) and mean (± s.e.m., B, C and E) data for SHR (open symbols) and WKY (filled symbols) at 37 °C and 1 Hz stimulation frequency. A, examples of raw data recorded for three consecutive cardiac cycles in 1.5 mm[Ca2+]o for representative WKY (left) and SHR (right) trabeculae. B and C, SHR (n = 6) and WKY (n = 5 for 4.0 mm[Ca2+]o, n = 4 for 5.0 mm[Ca2+]o, n = 6 otherwise). B, peak systolic (squares) and diastolic (circles) stress as a function of[Ca2+]o. Diastolic stress did not vary with either[Ca2+]o or rat strain. Peak systolic stress increased with increasing[Ca2+]o between 0.5 and 2.5 mm in WKY, and between 0.5 and 3.5 mm in SHR, but was reduced in SHR when compared to WKY for all[Ca2+]o above 0.5 mm (P≤ 0.05 for the interaction between rat strain and[Ca2+]o). C, peak systolic and resting [Ca2+]i as a function of[Ca2+]o. Resting [Ca2+]i (circles) did not change with[Ca2+]o, apart from at 5 mm, for either SHR or WKY, but was elevated in SHR (open circles) compared to WKY (filled circles) over all[Ca2+]o (P≤ 0.05). Peak systolic [Ca2+]i (squares) increased with increasing[Ca2+]o for all[Ca2+]o in SHR, but only between 0.5 and 2.0 mm in WKY, and was higher in SHR when compared to WKY for all[Ca2+]o (P≤ 0.05). D, data from typical experiments shown as phase plots for two different[Ca2+]o: an intermediary concentration (WKY: 1.5 mm; SHR: 3.5 mm), and the[Ca2+]o at which force was maximal for these trabeculae (WKY: 3.5 mm; SHR: 5 mm). E, peak systolic stress (from B) plotted as a function of peak systolic [Ca2+]i (from C).
Figure 3. The effect of[Ca2+]o on the time constant of fluorescence decay.
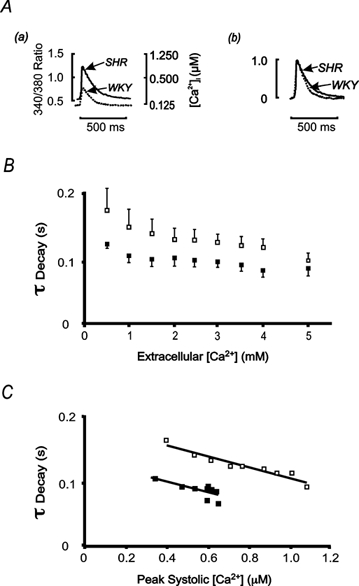
A, averaged fluorescence transients from representative SHR (continuous line) and WKY (dotted line) trabeculae at 1 Hz stimulation frequency (2 mm[Ca2+]o, 37 °C). In panel a both the 340 nm/380 nm fluorescence ratio and the corresponding [Ca2+]i are shown. Panel b shows the same fluorescence transients as in a normalised to their respective peak values. B, the time constant of decay (τDecay) of fluorescence decreased with increasing[Ca2+]o for both SHR (open symbols) and WKY (filled symbols), but was greater in SHR over all[Ca2+]o (P≤ 0.05). Data are means ± s.e.m. from SHR (n = 6) and WKY (n = 5 for 4.0 mm[Ca2+]o, n = 4 for 5.0 mm[Ca2+]o, n = 6 otherwise). C, mean data for the time constant of decay (as in B above) of fluorescence ratio (parameter A1 in eqn (2)) as a function of peak systolic [Ca2+]i. No difference of slopes of linear regression relations between SHR (y =−0.088x+ 0.19, R2= 0.91) and WKY (y =−0.084x+ 0.13, R2= 0.63) was found. Error bars have been omitted for clarity.
Figure 2B shows the[Ca2+]o dependence of resting and peak stress. Over the range 0.5 to 5.0 mm[Ca2+]o, resting stress was constant and did not differ between rat strains. However, the inotropic response to increasing[Ca2+]o was quite different between strains. The maximum active twitch stress was 63 % higher in WKY than in SHR, and occurred at a lower[Ca2+]o. No difference was detected in the time course of mechanical relaxation between SHR and WKY (data not shown). Figure 2C shows the[Ca2+]o dependence of peak and resting [Ca2+]i. Resting [Ca2+]i increased with increasing[Ca2+]o for both SHR and WKY (P ≤ 0.0001), and was greater in SHR compared to WKY at all[Ca2+]o examined (P ≤ 0.05). Peak systolic [Ca2+]i was also higher in SHR than in WKY at each[Ca2+]o. The effect of[Ca2+]o on peak systolic [Ca2+]i was different between rat strains (P ≤ 0.0001). While the amplitude of the Ca2+ transient (i.e. the difference between the peak and the resting [Ca2+]i) increased monotonically with[Ca2+]o in SHR trabeculae, it was significantly depressed at the highest concentration examined (5 mm) in the WKY preparations (n = 4). In addition, there was no increase in the amplitude of the [Ca2+]i transient in WKY for[Ca2+]o greater than 2 mm. Thus, while the[Ca2+]o dependence of the changes in force followed those of [Ca2+]i, the paradoxical reduction in SHR force production (given a larger peak [Ca2+]i) was observed at all[Ca2+]o. These marked differences in the relationship between stress and [Ca2+]i between strains is emphasised in the phase-plane plots from representative trabeculae shown in Fig. 2D. This figure shows that, during the complete contractile cycle, for a range of[Ca2+]o, SHR trabeculae were less responsive to [Ca2+]i than were WKY trabeculae. This view is reinforced by the data in Fig. 2E where peak stress development is plotted as a function of the peak [Ca2+]i. It is notable that there is no overlap between the relationships for WKY and SHR.
Although the time-to-peak fluorescence was not affected by altering[Ca2+]o, for either strain, the maximum rate of rise of fluorescence increased similarly with[Ca2+]o in both SHR and WKY (data not shown). The reduced rate of decay of the Ca2+ transient described above was also observed as[Ca2+]o was varied. Representative records of fluorescence are shown in Fig. 3Aa, and are normalised to their peak amplitudes in Fig. 3Ab. The slower rate of decay of the Ca2+ transient in SHR trabeculae was observed at all[Ca2+]o (Fig. 3B). Since the peak [Ca2+]i was generally higher in SHR than in WKY, the reduced rate of decay of the fluorescence transient might be related to the non-linear nature of the fura-2 response (since dF/d[Ca2+] decreases with increasing [Ca2+]). That this was not the case is suggested by the data shown in Fig. 3C, which shows that, regardless of peak [Ca2+]i, the time constant of [Ca2+]i decay is larger in SHR than in WKY trabeculae. Since the rate of decay of the Ca2+ transient is dominated by the activity of the SR Ca2+ pump, these data suggest that SR content might be reduced in SHR which, in turn, would be expected to reduce the amplitude of the Ca2+ transient (Janczewski & Lakatta, 1993). This is clearly not the case with our data, suggesting that the increased amplitude of the Ca2+ transient must be related to the elevated resting [Ca2+]i rather than the activity of the SR Ca2+-ATPase per se. Since the resting level of [Ca2+]i is set by SL Ca2+ transport, we carried out experiments to look for differences in SL Ca2+ transport between SHR and WKY.
Dissection of sarcolemmal transport mechanisms
Prolonged application of caffeine was used to discharge the SR Ca2+ store, and functionally eliminate its contribution to the decay in [Ca2+]i. The time course of the decay in the subsequent caffeine-evoked Ca2+ transient was then used to probe the relative contributions of the Na+-Ca2+ exchanger and the SL Ca2+-ATPase. Figure 4A shows the time course of fluorescence change in a representative experiment in which both the Na+-Ca2+ exchanger and the SL Ca2+-ATPase were active. After removing extracellular Ca2+, application of 10 mm caffeine caused an increase in [Ca2+]i which declined in ≈30 s. Figure 4B shows that after removal of Ca2+ and Na+, and application of Ni+, the caffeine-evoked increase in Ca2+ took more than three times as long to decay. Comparison of Fig. 4A with B after removal of caffeine and return to control solutions also shows that, when the Na+-Ca2+ exchanger was not blocked (Fig. 4A), a marked increase in resting [Ca2+]i developed – an effect that might be related to Na+ accumulation during extrusion of Ca2+ via the Na+-Ca2+ exchange mechanism. The onset of stimulation following caffeine exposure did not immediately elicit SR Ca2+ release, indicating that SR Ca2+ stores had indeed been discharged by exposure to 10 mm caffeine.
Figure 4. Contribution of sarcolemmal transport mechanisms to decay of [Ca2+]i.
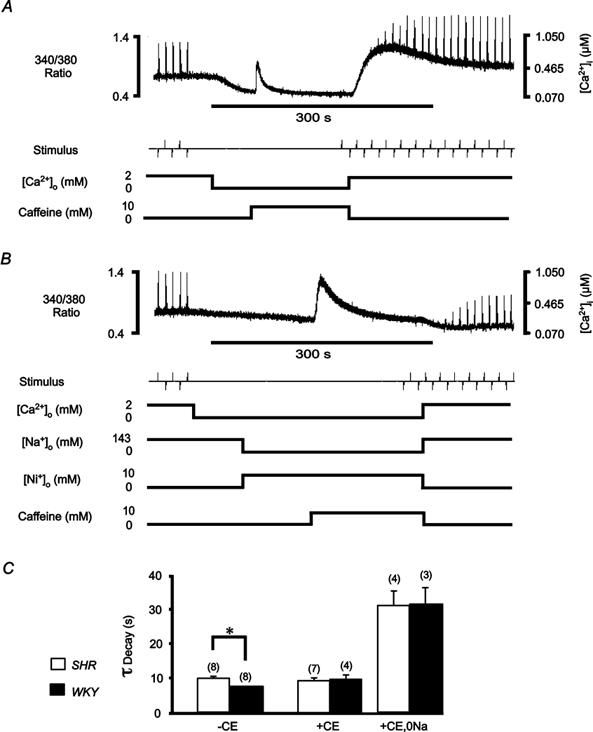
Quiescent trabeculae were exposed to 10 mm caffeine (0 mm[Ca2+]o, 22 °C) in order to functionally eliminate the SR Ca2+-ATPase. Trabeculae were stimulated at 0.1 Hz (2 mm[Ca2+]o) before and after caffeine application. An exponential function was fitted to the decay of the fluorescence ratio, from the peak of the caffeine transient, in order to determine the time constant. A, continuous recording (500 s) of the emitted 340 nm/380 nm fluorescence ratio before, during and after caffeine application in a representative LV trabecula from SHR. Following withdrawal of caffeine, the fluorescence ratio indicates a large increase in the resting [Ca2+]i that slowly decreases back to the pre-caffeine level (complete recovery not shown). B, continuous recording of the fluorescence ratio before, during and after application of caffeine in the same trabecula as in A, subsequent to loading with carboxyeosin (CE), and in the presence of 10 mm Ni+ and 0 mm Na+. Note that on withdrawal of caffeine, the fluorescence ratio shows no rapid increase in resting [Ca2+]i, suggesting that the Na+-Ca2+ exchanger was completely blocked during the caffeine application, in contrast to A. C, mean ± s.e.m. time constants (τ) of fluorescence decay during caffeine application in 0 mm Ca2+ solution (143 mm Na+) before (-CE) and after (+CE) inhibiting the SL Ca2+-ATPase and after blocking both SL transport mechanisms (+CE, 0 Na). Numbers of trabeculae as indicated in parentheses. *P < 0.05.
Results of similar experiments are summarised in Fig. 4C. The time constant of the decay in [Ca2+]i in the presence of 10 mm caffeine was greater for SHR trabeculae than for WKY trabeculae (9.8 ± 0.7 s (n = 8) vs. 7.7 ± 0.2 s (n = 8), respectively). When CE was applied to block the SL Ca2+-ATPase this difference between SHR and WKY was abolished, such that the WKY transient became as slow as that of the SHR (9.3 ± 0.9 s (n = 7)vs. 9.7 ± 1.4 s (n = 4), SHR vs. WKY, respectively). Since the resting [Ca2+]i was increased for all SHR trabeculae (mean increase 124 %, n = 5, P ≤ 0.05) following CE loading, as was peak fluorescence for electrically evoked transients for both SHR and WKY (P ≤ 0.05), it was evident that a 30 min CE loading period was sufficient for both rat strains. No effect of CE on Ca2+-independent fluorescence was found following washout. As might be expected, after inhibiting both the Na+-Ca2+ exchanger and the SL Ca2+-ATPase, the rate of decay of [Ca2+]i was significantly slowed, but with no difference in the time course of decay apparent between SHR and WKY (31.0 ± 4.4 s (n = 4)vs. 31.6 ± 4.7 s (n = 3), respectively). In these conditions, the amplitude of the caffeine-evoked Ca2+ transient was also larger in SHR (0.40 ± 0.13 vs. 0.19 ± 0.04 μm in WKY), a result consistent with the conclusion reached above that the SR calcium content (and hence release) is larger in SHR. These data confirm that the origin of the difference in decline of the caffeine-evoked Ca2+ transient resides in SL Ca2+ transport. Taken together, these observations suggest that in SHR the Ca2+ extrusion capacity of the SL Ca2+-ATPase was reduced compared to WKY. While such a difference can explain the increased resting [Ca2+]i, and hence the increased Ca2+ transient amplitude due to increased SR Ca2+ content, it does not explain the reduced contractile force in SHR. Since force development may be impeded by components of the extracellular matrix (Brilla et al. 1996), we examined the possibility that a proliferation of collagen might impede the contraction of SHR trabeculae.
Distribution and content of collagen in trabeculae
Transverse sections of trabeculae were stained (see Methods) to allow examination of collagen within trabeculae. Figure 5 shows data from representative WKY and SHR LV trabeculae of similar diameter. Panels A and D show that, despite comparable fluorescence records, the SHR preparation contracted much more weakly. Cross-sections of the trabeculae are shown in Fig. 5B and E, where the pink-stained collagen fibres are seen to be more prevalent in the SHR. Analysis of cross-sections showed that average collagen was sixfold higher in LV trabeculae from SHR (18.0 ± 4.6 % (n = 10)vs. 2.9 ± 0.9 % (n = 11) in WKY). The longitudinal distribution of collagen can be seen in Fig. 5C and F. The difference in collagen organisation was marked; in WKY the collagen generally formed quite uniform longitudinal bands (aligned in parallel with the myocytes), whereas in SHR the collagen was far more branched with many strands running transversely, forming a mesh-like structure. Since such a mesh would inhibit the ability of cells to shorten at constant volume, these data indicate that matrix remodelling during the progression to heart failure may be a major contributory factor to failure of the myocytes to develop full force, despite the availability of adequate calcium. To examine this point further, we normalised force to the proportion of cross-sectional area occupied by myocytes (rather than to total cross-sectional area). When this correction was applied, the difference in force production at 5 Hz stimulation frequency remained reduced for SHR, peak systolic stress (8.1 ± 1.8 vs. 22.3 ± 5.6 mN mm−2 of myocyte), with no difference in diastolic stress (4.4 ± 1.4 vs. 3.6 ± 2.3 mN mm−2 of myocyte) between SHR (n = 4) and WKY (n = 5), respectively. In other words, it cannot be the collagen per se which is responsible for diminished contractile performance, but rather the complex three-dimensional structure of the collagen.
Discussion
The spontaneously hypertensive rat (SHR) is a widely studied animal model of systemic hypertension that progresses to heart failure in a manner which has similarities to the human disease. However, although widely studied, it remains unclear whether abnormal myocardial calcium transport plays a role in the genesis of end-stage contractile failure in this animal model, since few studies have examined both force and [Ca2+]i in failing SHR hearts. In this study, LV trabeculae from SHR in heart failure produced only 40 % of the peak systolic stress of age-matched WKY, but paradoxically, the amplitude of the Ca2+ transient was larger in SHR than in WKY. These results were unexpected, since a number of studies have reported reduced contractility in heart failure in conjunction with diminished Ca2+ transients (Beuckelmann et al. 1992; Pieske et al. 1995; Gómez et al. 1997). However, it should be noted that this is the first study that has employed ratiometric indicators in a contracting cardiac muscle preparation at physiological temperature and stimulation frequency. It is also notable that we were able to identify a sarcolemmal origin for the increased amplitude of the Ca2+ transient, reflecting the contribution of SL Ca2+ transport to SR Ca2+ content (for review see Eisner et al. 1998). Nevertheless, our data are in accord with studies that show decreased rate of SR uptake, the difference being that this decrease is offset by an increased resting [Ca2+]i level (which would allow the SR to accumulate more calcium despite a reduction in the rate of uptake).
Given that both our data and previous reports (Bing et al. 1991; Brooks et al. 1994; Gómez et al. 1997) show reduced contractile performance for SHR in heart failure, and the well-known relationship between Ca2+ and force, how can our observation of reduced twitch force in the face of an increased Ca2+ transient be explained? The simplest explanation would be a decrease in the sensitivity of the contractile machinery to [Ca2+]i, which would also explain a slight increase in the peak amplitude of the Ca2+ transient (Allen & Kentish, 1985). However, such a decrease in Ca2+ sensitivity is unlikely, since for experiments carried out at high[Ca2+]o, whilst [Ca2+]i continued to increase in SHR trabeculae, force reached a plateau (Fig. 2B and E). Furthermore, Perreault et al. (1990) found no difference in the myofilament Ca2+ responsiveness in skinned LV muscles from SHR with hypertrophy and heart failure. Although myosin isoform changes have been reported (Lompre et al. 1979; Bing et al. 1991; Boluyt et al. 1994), our data do not support the idea that the contractile deficit resides in the rate of cross-bridge cycling per se, since the normalised rate of rise of force (normalised to peak stress) was only 10 % greater in WKY than in SHR. This view is in accord with measurements of cross-bridge cycling in a pressure overload model, which suggested only minor changes in cross-bridge function (Peterson et al. 2001). Although the time constant of decay in [Ca2+]i was larger for SHR than for WKY, we found no difference in the rate of relaxation of force. This result is consistent with a recent report by Janssen et al. (2002) who showed that under experimental conditions resembling those that prevail in vivo, and as used here, relaxation rate was limited by cross-bridge cycling. Since the relaxation rate was unaltered in SHR, these data further argue against impaired force production residing in altered cross-bridge cycling.
If reduced force production is not explainable on the basis of a reduced Ca2+ transient, reduced myofilament Ca2+ sensitivity, or reduced cross-bridge cycling, then we must look beyond the force generating machinery for an explanation. Increases in myocardial fibrosis and passive stiffness have previously been reported in failing hearts from SHR (Conrad et al. 1995) as well as increases in the expression of genes encoding extracellular matrix (Boluyt et al. 1994; Bing et al. 1997; Boluyt & Bing, 2000). In accord with these data, we found increased collagen bundles within the SHR trabeculae, which were more densely interwoven. When myocytes contract, constant volume behaviour requires an increase in cross-section concomitant with shortening. (Although the contractions of our trabeculae are nominally isometric, the inevitable compliance at the ends of the preparations would result in myocyte shortening of about 12 % (ter Keurs et al. 1980).) While the longitudinal arrangement of collagen in WKY would hardly resist such increases in myocyte cross-section, this would not be the case in SHR where the extensive collagen network might well impede cell shortening.
Comparison to previous studies
In our study we sought to examine the behaviour of LV trabeculae under minimally perturbing conditions, an aim which could not be attained by employing voltage clamp experiments on isolated myocytes (e.g. Gómez et al. 1997) or in large multicellular preparations such as LV papillary muscles. LV trabeculae are much thinner preparations than papillary muscles, which should avoid metabolic problems associated with excessive diffusion distances (Schouten & ter Keurs, 1986; Janssen et al. 2002). The mean diameter of LV trabeculae used in our study was ≈200 μm and trabeculae of this diameter have been shown to produce maximal force at a stimulation frequency of ≈6 Hz (Janssen et al. 2002) compatible with the in vivo rate chosen here. That such preparations are not metabolically compromised has been shown by Daut & Elzinga (1988). In any case, we also observed reduced force in all SHR preparations at lower frequencies and lower bathing Ca2+ (see Fig. 2), which would rule out an explanation based simply on compromised metabolism within our trabeculae.
Bing et al. (1991) measured [Ca2+]i and found that there were significant differences between failing SHR and their controls. They reported that the time-to-peak aequorin luminescence was prolonged in failing SHR papillary muscles, and that resting [Ca2+]i was lower (Bing et al. 1991). These results differ from our measurements in LV trabeculae made using fura-2, but direct comparison is confounded by the non-linearity of the aequorin response at low Ca2+ concentrations (Allen et al. 1977), the lower temperature, and the lower stimulation frequencies used in the study of Bing et al. (1991). However, a lower stimulation rate does not simply explain the difference between their results and those reported here, since, even at 1 Hz, we observed a larger Ca2+ transient in SHR (see Fig. 2B and D). Furthermore, in partial agreement with our data, Shorofsky et al. (1999) reported that the amplitudes of both Ca2+ transients and Ca2+ sparks were larger in hypertrophied myocytes from young adult SHR. Engelmann et al. (1987) reported that the volume fraction of myocytes is decreased in senescent SHR ventricles compared to age-matched WKY, and we should expect the fluorescence of interstitial cells to contribute to resting [Ca2+]i measurements in fura-2 AM-loaded trabeculae. However, the loss of myocyte volume is small (< 10 %) compared to the 50-110 % increase in resting [Ca2+]i observed, which seems inconsistent with the changes in resting [Ca2+]i being due to a differential increase in non-myocyte cell volume. It should also be noted that, without an increase in resting [Ca2+]i, an increase in Ca2+ transient amplitude in the presence of a small decrease in SR uptake rate (estimated from the rate of fluorescence) is problematic (see below).
Kinetics of the Ca2+ transient
We found that the decay of the Ca2+ transient was slower for SHR over a range of different experimental conditions, as has been previously reported in many models of hypertrophy and heart failure. This slowing is most frequently attributed to a reduced rate of SR Ca2+-ATPase activity (for a review see Movsesian & Schwinger, 1998). Although a reduced rate of SR Ca2+-ATPase activity might contribute to the slower decay of the Ca2+ transient in our experiments, it is important to note that this does not appear to lead to a reduced SR content (as judged by the amplitudes of the Ca2+ transient and caffeine-evoked SR release). We suggest that the reduced rate of SR uptake is offset by an increased resting [Ca2+]i which permits the SR to accumulate more Ca2+ during the period between stimuli. In connection with this point, it is noteworthy that while the SR uptake rate of SHR was reduced to ≈60 % of WKY (see Fig. 3C), the resting level of [Ca2+]i was increased by ≈55 % (see Fig. 2C). This increase in resting [Ca2+]i would be sufficient to offset the reduction in SR uptake rate if the net SR calcium accumulation during diastole were (for example) proportional to the uptake rate times the resting level of [Ca2+]i.
The decreased rate of SR uptake may not be related to SR Ca2+-ATPase levels per se since Northern blot analysis did not show a significant difference in the level of the mRNA between WKY and SHR in heart failure (Boluyt et al. 1994). An alternative explanation would be that a prolongation of membrane depolarisation delays the return of Ca2+ to resting levels (Cannell et al. 1987). In support of this suggestion, electrophysiological studies have shown a longer action potential duration with hypertrophy and heart failure in SHR (Brooksby et al. 1993; Cerbai et al. 1994). It should be noted that increased action potential duration also increases the SR Ca2+ content (Terracciano et al. 1997).
Diastolic Ca2+
The trans-sarcolemmal Ca2+ transporters, the Na+-Ca2+-exchanger and the Ca2+-ATPase, are the primary determinants of resting [Ca2+]i, with the former being the more important (Crespo et al. 1990; Lamont & Eisner, 1996; Choi et al. 2000). Since Na+-Ca2+ exchange is very sensitive to the resting level of [Na+]i (Eisner & Lederer, 1985), an increase of [Na+]i could explain the elevated steady-state [Ca2+]i. Such an increase might arise either from an increase in action potential duration (see above) or from a decrease in Na+-K+-ATPase activity. Some support for the latter possibility has been obtained in human heart failure where decreased expression of the Na+-K+-ATPase has been observed (Schwinger et al. 1999). However, our experiments with caffeine suggest that the SL Ca2+-ATPase may also be implicated since the carboxyeosin-sensitive extrusion was 22 % lower in SHR than in WKY (see Fig. 4C). Of course, the relative importance of this reduction is predicated on the relative importance of the SL Ca2+-ATPase Ca2+ extrusion pathway compared to that of Na+-Ca2+ exchange. If the SL Ca2+-ATPase is a minor component of resting calcium extrusion at body temperature, then it follows that the significant increase in resting [Ca2+]i observed here probably resides in alterations in Na+-Ca2+ exchange. Finally, differences in SL transport between SHR and WKY may merely reflect the increased surface to volume ratio of hypertrophic myocytes, rather than a difference of either protein expression levels, or specific activity. Nevertheless, the observed increase in resting [Ca2+]i in SHR trabeculae must be due to changes in SL Ca2+ handling.
In summary, we have found that at 37 °C, and at a stimulation frequency approximating that found in vivo, LV trabeculae from failing SHR hearts demonstrate an elevated resting [Ca2+]i and an increased peak systolic [Ca2+]i, but a decreased peak systolic stress. Our results therefore show that the reduced contractile response of LV trabeculae from SHR in heart failure is not due to reduced myofilament Ca2+ availability. Morphological examination of trabeculae showed that the collagen content is increased in SHR compared to WKY, and that the longitudinal distribution contains extensive branching. In SHR, the progression from stable hypertrophy to heart failure occurs slowly over a period of months, unlike many other experimental models. We suggest that the observed alterations in the Ca2+ transient may have developed in the SHR as a compensatory response to changes in the extracellular matrix that compromise contractility.
Acknowledgments
This work was supported by the National Heart Foundation of New Zealand. Marie-Louise Ward was the recipient of an Auckland Medical Research Foundation Senior Scholarship. Confocal microscopy and digital imaging were carried out with the assistance of Hilary Holloway at the University of Auckland Biomedical Imaging Research Unit. Cardiac ultrasound was carried out by Helen Walsh and Gillian Whalley, Department of Medicine, University of Auckland.
References
- Adams W, Trafford AW, Eisner DA. 2,3-Butanedione monoxime (BDM) decreases sarcoplasmic reticulum Ca content by stimulating Ca release in isolated rat ventricular myocytes. Pflugers Arch. 1998;436:776–781. doi: 10.1007/s004240050701. [DOI] [PubMed] [Google Scholar]
- Allen DG, Blinks JR, Prendergast FG. Aequorin luminescence: relation of light emission to calcium concentration – a calcium-independent component. Science. 1977;195:996–998. doi: 10.1126/science.841325. [DOI] [PubMed] [Google Scholar]
- Allen DG, Jewell BR, Wood EH. Studies of the contractility of mammalian myocardium at low rates of stimulation. J Physiol. 1976;254:1–17. doi: 10.1113/jphysiol.1976.sp011217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Kentish JC. The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- Assayag P, Charlemagne D, De Leiris J, Boucher F, Valere P-E, Lortet S, Swynghedauw B, Besse S. Senescent heart compared with pressure overload-induced hypertrophy. Hypertension. 1997;29:15–21. doi: 10.1161/01.hyp.29.1.15. [DOI] [PubMed] [Google Scholar]
- Aubin JE. Autofluorescence of viable cultured mammalian cells. J Histochem Cytochem. 1979;27:36–43. doi: 10.1177/27.1.220325. [DOI] [PubMed] [Google Scholar]
- Bakker AJ, Head SI, Williams DA, Stephenson DG. Ca2+ levels in myotubes grown from the skeletal muscle of dystrophic (mdx) and normal mice. J Physiol. 1993;460:1–13. doi: 10.1113/jphysiol.1993.sp019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. SR Ca loading in cardiac muscle preparations based on rapid cooling contractures. Am J Physiol. 1989;256:C109–120. doi: 10.1152/ajpcell.1989.256.1.C109. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- Bing OHL, Brooks WW, Conrad CH, Sen S, Perreault CL, Morgan JP. Intracellular calcium transients in myocardium from spontaneously hypertensive rats during the transition to heart failure. Circ Res. 1991;68:1390–1400. doi: 10.1161/01.res.68.5.1390. [DOI] [PubMed] [Google Scholar]
- Bing OHL, Brooks WW, Robinson KG, Slawsky MT, Hayes JA, Litwin SE, Sen S, Conrad CH. The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. J Mol Cell Cardiol. 1995;27:383–396. doi: 10.1016/s0022-2828(08)80035-1. [DOI] [PubMed] [Google Scholar]
- Bing OHL, Ngo HQ, Humphries DE, Robinson KG, Lucey EC, Carver W, Brooks WW, Conrad CH, Hayes JA, Goldstein RH. Localization of α1(I) collagen mRNA in myocardium from the spontaneously hypertensive rat during the transition from compensated hypertrophy to failure. J Mol Cell Cardiol. 1997;29:2335–2344. doi: 10.1006/jmcc.1997.0465. [DOI] [PubMed] [Google Scholar]
- Boluyt MO, Bing OHL. Matrix gene expression and decompensated heart failure: the aged SHR model. Cardiovasc Res. 2000;46:239–249. doi: 10.1016/s0008-6363(00)00043-2. [DOI] [PubMed] [Google Scholar]
- Boluyt MO, O'Neill L, Meredith AL, Bing OHL, Brooks WW, Conrad CH, Crow MT, Lakatta EG. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure: marked upregulation of genes encoding extracellular matrix components. Circ Res. 1994;75:23–32. doi: 10.1161/01.res.75.1.23. [DOI] [PubMed] [Google Scholar]
- Brandes R, Bers DM. Increased work in cardiac trabeculae causes decreased mitochondrial NADH fluorescence followed by slow recovery. Biophys J. 1996;71:1024–1035. doi: 10.1016/S0006-3495(96)79303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilla CG, Matsubara L, Weber KT. Advanced hypertensive heart disease in spontaneously hypertensive rats. Hypertension. 1996;28:269–275. doi: 10.1161/01.hyp.28.2.269. [DOI] [PubMed] [Google Scholar]
- Brooks WW, Bing HL, Litwin SE, Conard CH, Morgan JP. Effects of treppe and calcium on intracellular calcium and function in the failing heart from the spontaneously hypertensive rat. Hypertension. 1994;24:347–356. doi: 10.1161/01.hyp.24.3.347. [DOI] [PubMed] [Google Scholar]
- Brooksby P, Levi AJ, Jones JV. The electrophysiological characteristics of hypertrophied ventricular myocytes from the spontaneously hypertensive rat. J Hypertension. 1993;11:611–622. doi: 10.1097/00004872-199306000-00005. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Barbieri M, Li Q, Mugelli A. Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovasc Res. 1994;28:1180–1187. doi: 10.1093/cvr/28.8.1180. [DOI] [PubMed] [Google Scholar]
- Choi HS, Trafford AW, Eisner DA. Measurement of calcium entry and exit in quiescent rat ventricular myocytes. Pflugers Arch. 2000;440:600–608. doi: 10.1007/s004240000295. [DOI] [PubMed] [Google Scholar]
- Choi JS, Eisner DA. The role of sarcolemmal Ca2+-ATPase in the regulation of resting calcium concentration in rat ventricular myocytes. J Physiol. 1999;515:109–118. doi: 10.1111/j.1469-7793.1999.109ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OHL. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161–170. doi: 10.1161/01.cir.91.1.161. [DOI] [PubMed] [Google Scholar]
- Conrad CH, Brooks WW, Robinson KG, Bing OHL. Impaired myocardial function in spontaneously hypertensive rats with heart failure. Am J Physiol. 1991;260:H136–145. doi: 10.1152/ajpheart.1991.260.1.H136. [DOI] [PubMed] [Google Scholar]
- Crespo LM, Grantham CJ, Cannell MB. Kinetics, stoichiometry and role of the Na-Ca exchange mechanism in isolated cardiac myocytes. Nature. 1990;345:618–621. doi: 10.1038/345618a0. [DOI] [PubMed] [Google Scholar]
- Daut J, Elzinga G. Heat production of quiescent ventricular trabeculae isolated from guinea-pig heart. J Physiol. 1988;398:259–275. doi: 10.1113/jphysiol.1988.sp017041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Steinberg TH, Siverstein SC. Inhibition of fura-2 sequestration and secretion with organic anion transport blockers. Cell Calcium. 1990;11:57–62. doi: 10.1016/0143-4160(90)90059-4. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Lederer WJ. Na-Ca exchange: stoichiometry and electrogenicity. Am J Physiol. 1985;248:C189–202. doi: 10.1152/ajpcell.1985.248.3.C189. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Trafford AW, Diaz ME, Overend CL, O'Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- Engelmann GL, Vitullo JC, Gerrity RG. Morphometric analysis of cardiac hypertrophy during development, maturation, and senescence in spontaneously hypertensive rats. Circ Res. 1987;60:487–494. doi: 10.1161/01.res.60.4.487. [DOI] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Moss RL. Aging-dependent depression in the kinetics of force development in rat skinned myocardium. Am J Physiol. 1999;276:H1511–1519. doi: 10.1152/ajpheart.1999.276.5.H1511. [DOI] [PubMed] [Google Scholar]
- Gómez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Loiselle DS. Mechanisms of force inhibition by halothane and isoflurane in intact rat cardiac muscle. J Physiol. 1998;506:231–244. doi: 10.1111/j.1469-7793.1998.231bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Young AA, Legrice IJ, Edgar SG, Loiselle DS. 3-Dimensional configuration of perimysial collagen fibres in rat cardiac muscle at resting and extended sarcomere lengths. J Physiol. 1999;517:831–837. doi: 10.1111/j.1469-7793.1999.0831s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RA, Redon D. Calibration of intracellular Ca transients of isolated adult heart cells labelled with fura-2 by acetoxymethyl ester loading. Cell Calcium. 1998;24:263–273. doi: 10.1016/s0143-4160(98)90050-1. [DOI] [PubMed] [Google Scholar]
- Janczewski AM, Lakatta EG. Thapsigargin inhibits Ca2+ uptake, and Ca2+ depletes sarcoplasmic reticulum in intact cardiac myocytes. Am J Physiol. 1993;265:H517–522. doi: 10.1152/ajpheart.1993.265.2.H517. [DOI] [PubMed] [Google Scholar]
- Janssen PML, Stull LB, Marban E. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am J Physiol Heart Circ Physiol. 2002;282:H499–507. doi: 10.1152/ajpheart.00595.2001. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Julian FJ. Pacing rate, halothane, and BDM affect fura 2 reporting of [Ca2+]i in intact rat trabeculae. Am J Physiol. 1997;273:C2046–2056. doi: 10.1152/ajpcell.1997.273.6.C2046. [DOI] [PubMed] [Google Scholar]
- Lamont C, Eisner DA. The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflugers Arch. 1996;432:961–969. doi: 10.1007/s004240050223. [DOI] [PubMed] [Google Scholar]
- Lompre A-M, Schwartz K, d’Albis A, Lacombe G, Van Thiem N, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature. 1979;282:105–107. doi: 10.1038/282105a0. [DOI] [PubMed] [Google Scholar]
- Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. Contribution of reverse-mode sodium-calcium exchange to contractions in failing human left ventricular myocytes. Cardiovasc Res. 1998;37:424–431. doi: 10.1016/s0008-6363(97)00271-x. [DOI] [PubMed] [Google Scholar]
- Movsesian MA, Schwinger RHG. Calcium sequestration by the sarcoplasmic reticulum in heart failure. Cardiovasc Res. 1998;37:352–359. doi: 10.1016/s0008-6363(97)00259-9. [DOI] [PubMed] [Google Scholar]
- Mulieri LA, Hasenfuss G, Ittleman F, Blanchard EM, Alpert NR. Protection of human left ventricular myocardium from cutting injury with 2,3-butanedione monoxime. Circ Res. 1989;65:1441–1444. doi: 10.1161/01.res.65.5.1441. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jap Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- O'Neill SC, Eisner DA. A mechanism for the effects of caffeine on Ca2+ release during diastole and systole in isolated rat ventricular myocytes. J Physiol. 1990;430:519–536. doi: 10.1113/jphysiol.1990.sp018305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault CL, Bing OHL, Brooks WW, Ransil BJ, Morgan JP. Differential effects of cardiac hypertrophy and failure on right versus left ventricular calcium activation. Circ Res. 1990;67:707–712. doi: 10.1161/01.res.67.3.707. [DOI] [PubMed] [Google Scholar]
- Peterson JN, Nasser R, Anderson PAW, Alpert NR. Altered cross-bridge characteristics following haemodynamic overload in rabbit hearts expressing V3 myosin. J Physiol. 2001;536:569–582. doi: 10.1111/j.1469-7793.2001.0569c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- Rodriguez EK, Hunter WC, Royce MJ, Leppo MK, Douglas AS, Weisman HF. A method to reconstruct myocardial sarcomere lengths and orientations at transmural sites in beating canine hearts. Am J Physiol. 1992;263:H293–306. doi: 10.1152/ajpheart.1992.263.1.H293. [DOI] [PubMed] [Google Scholar]
- Schouten VJA, ter Keurs HEDJ. The force-frequency relationship in rat myocardium: the influence of muscle dimensions. Pflugers Arch. 1986;407:14–17. doi: 10.1007/BF00580714. [DOI] [PubMed] [Google Scholar]
- Schwinger RHG, Wang J, Frank K, Muller-Ehmsen J, Brixius K, McDonough AA, Erdmann E. Reduced sodium pump α1, α3 and β1-isoform protein levels but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999;99:2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- Shorofsky SR, Aggarwal R, Corretti M, Baffa J, Strum JM, Al-Seikhan BA, Kobayashi YM, Jones LR, Wier WG, Balke CW. Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks. Circ Res. 1998;84:424–434. doi: 10.1161/01.res.84.4.424. [DOI] [PubMed] [Google Scholar]
- Steele JR, Smith GL. Effects of 2,3-butanedione monoxime on sarcoplasmic reticulum of saponin-treated rat cardiac muscle. Am J Physiol. 1993;265:H1493–1500. doi: 10.1152/ajpheart.1993.265.5.H1493. [DOI] [PubMed] [Google Scholar]
- ter Keurs HEDJ, Rijnsburger WH, Van Heuningen R, Nagelsmit MJ. Tension development and sarcomere length in rat cardiac trabeculae: evidence of length-dependent activation. Circ Res. 1980;46:703–714. doi: 10.1161/01.res.46.5.703. [DOI] [PubMed] [Google Scholar]
- Terracciano CMN, Tweedie D, MacLeod KT. The effects of changes to action potential duration on the calcium content of the sarcoplasmic reticulum in isolated guinea-pig ventricular myocytes. Pflugers Arch. 1997;433:542–544. doi: 10.1007/s004240050312. [DOI] [PubMed] [Google Scholar]